


Генетичні захворюванняСерповидно-клітинна анемія (частина перша) Серповидно-клітинна хвороба (СКХ) або серповидно-клітинна анемія (або анемія; СКА), або дрепаноцитоз - це аутосомно-рецесивне наддомінантне генетичне захворювання крові, яке характеризується неправильною, стійкою, серповидною формою червоних кров'яних клітин (еритроцитів). Серповидність клітин зменшує їх гнучкість та еластичність, що збільшує ризик виникнення різних ускладнень. Причиною появи клітин серповидної форми є мутації в гені гемоглобіну. Як наслідок скорочується очікувана тривалість життя, в середньому вона складає 42 роки у чоловіків і 48 у жінок.
Серповидно-клітинна анемія, як правило, проявляється в дитинстві, частіше зустрічається у людей (або їх нащадків) з тропічних та субтропічних регіонів, які є ендемічними, щодо малярії. Одна третина всіх корінних жителів Африки на південь від Сахари є носіями гену. У зв’язку з тим, що малярія є поширеним захворюванням у цьому регіоні носії одного гену хвороби володіють підвищеною стійкістю до виживання (серповидно-клітинна особливість). Ті, у кого є тільки одна з двох алелей серповидно-клітинної анемії, хоча є менш стійкими у них спостерігається більша стійкість організму до інфекції при зараженні.
За даними Національного інституту здоров’я поширеність цього захворювання в США складає приблизно 1 випадок на 5000 жителів, переважно ураженими є американці африканського походження з півдня Сахари. У Сполучених Штатах приблизно у однієї особи з 500 новонароджених негроїдної раси мають серповидно-клітинну анемію.
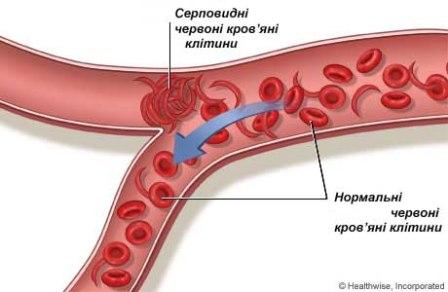 Дрепаноцитоз (серповидно-клітинна анемія) - назва специфічної форми серповидно-клітинної хвороби, при якій гомозиготність мутації спричиняє появу гемоглобіну S (HbS). Цей тип серповидно-клітинної анемії, також відомий як "HbSS", "SS хвороба", "гемоглобін S", дрепаноцитарна анемія, серповидноклітинна гемолітична анемія, африканська анемія, меніскоцитоз або синдром Херіка (Геріка). Гетерозигот, які мають тільки один серповидний ген та один нормальний ген гемоглобіну, називають "HbAS". Інші, більш рідкісні форми захворювання включають: серповидне захворювання гемоглобіну C (HbSC), серповидну бета-плюс (HbS / β+) та бета-нуль (HbS/β0) таласемії. Ці форми серповидно-клітинної анемії характеризуються явищем поєднаної гетерозиготності, при якій особа має тільки одну копію мутованого гена, який спричиняє HbS та одну копію іншої дефектної алелі гемоглобіну. Дрепаноцитоз (серповидно-клітинна анемія) - назва специфічної форми серповидно-клітинної хвороби, при якій гомозиготність мутації спричиняє появу гемоглобіну S (HbS). Цей тип серповидно-клітинної анемії, також відомий як "HbSS", "SS хвороба", "гемоглобін S", дрепаноцитарна анемія, серповидноклітинна гемолітична анемія, африканська анемія, меніскоцитоз або синдром Херіка (Геріка). Гетерозигот, які мають тільки один серповидний ген та один нормальний ген гемоглобіну, називають "HbAS". Інші, більш рідкісні форми захворювання включають: серповидне захворювання гемоглобіну C (HbSC), серповидну бета-плюс (HbS / β+) та бета-нуль (HbS/β0) таласемії. Ці форми серповидно-клітинної анемії характеризуються явищем поєднаної гетерозиготності, при якій особа має тільки одну копію мутованого гена, який спричиняє HbS та одну копію іншої дефектної алелі гемоглобіну. Термін захворювання застосовується, у зв’язку з тим, що саме успадкована аномалія є причиною патологічного стану, який може призвести до смерті і появи важких ускладнень. Не всі успадковані варіанти гемоглобіну є шкідливими, це явище відоме як генетичний поліморфізм.
Ознаки і симптоми
Серповидно-клітинне захворювання може призвести до виникнення різних гострих і хронічних ускладнень, деякі з них є потенційно смертельними. Серповидно-клітинна криза
Термін "серповидно-клітинна криза" використовується для опису декількох незалежних гострих станів, які зустрічаються у хворих. Серповидно-клітинна хвороба призводить до анемії з ймовірними різнотипними кризами, включаючи: судинно-оклюзійну, апластичну, секвестральну, гіпергемолітичну та інші кризи. Більшість епізодів серповидноклітинної кризи проявляються впродовж 5 - 7 днів. Судинно-оклюзійна криза
Судинно-оклюзійна криза викликана серповидною формою еритроцитів, які закупорюють капіляри і обмежують приплив крові до органу, спричиняючи ішемію, біль, некроз, а також часто пошкодження цього органу. Частота, тяжкість і тривалість цих криз суттєво відрізняються. Тяжкі кризи з больовими відчуттями вимагають лікування гідратацією, анальгетиками та переливанням крові. Для тамування болю необхідним є застосування опіатів через регулярні проміжки часу до нормалізації стану хворого. При більш легких формах криз, підгрупа пацієнтів може обмежитись прийомом нестероїдних протизапальних засобів (наприклад, диклофенаку або напроксену). При більш серйозних кризах для більшості хворих широко використовується стаціонарне лікування з внутрішньовенним введенням опіатів; контрольованою пацієнтом анальгезією (patient-controlled analgesia, або PCA). Інколи ефективним засобом для усунення свербіжу пов'язаного з вживання опіатів є димедрол. Прояви кризи у таких органах як пеніс та легені вимагають негайної госпіталізації та здійснення переливання крові. Рекомендованою є стимулювальна спірометрія – метод, при якому перевага надається глибокому диханню, завдяки якому ризик розвитку ателектазу зводиться до мінімуму. 
Криза секвестрації селезінки (Splenic sequestration crisis)
Через вузькі судини селезінки та виконання нею функції очищення (виведення дефектних еритроцитів),селезінка часто піддається ураженню. У осіб з серповидно-клітинною анемією інфаркт селезінки - є частим явищем в період дитинства. Автоспленектомія підвищує ризик зараження інкапсульованими організмами. При аспленії рекомендованими засобами є превентивні антибіотики та вакцинація. Криза секвестрації селезінки проявляється гострим, болючим збільшенням селезінки. Синусоїди та ворота селезінки, відкриваючись одночасно спричиняють раптовий приплив крові в селезінку та виникнення порушення кровообігу, що приводить до раптової гіповолемії. Живіт стає роздутим і дуже твердим. Секвестральна криза селезінки свідчить про критичний стан пацієнта. Якщо вчасно не почати лікування хворі можуть померти протягом 1-2 годин у зв’язку із гіповолемією і зниженням системного кров’яного тиску. Інколи лікування супроводжується переливанням крові. Ці кризи є швидкоплинними, минають за 3-4 години, а іноді можуть тривати добу. Апластична криза
Апластична криза є найбільш важким ускладненням фонової анемії у пацієнта, яка спричиняє блідість, тахікардію і втому. Цю кризу викликає парвовірус B19, який безпосередньо впливає на еритропоез (процес утворення червоних кров'яних тілець), та вторгається у прекурсори (попередники) червоних клітин, розмножуючись у них і знищуючи. Парвовірусна інфекція практично повністю пригнічує утворення еритроцитів впродовж двох-трьох днів. У здорових людей, цей процес не має особливих наслідків, проте у пацієнтів з серповидно-клітинною анемією тривалість життя еритроцитів знижується, тому ця ситуація є небезпечною для їх життя. Кількість ретикулоцитів (молодих незрілих еритроцитів, які містять ядра) під час хвороби різко падає (спричиняючи ретикулоцитопенію), а висока швидкість руйнування та утворення еритроцитів приводить до зниження гемоглобіну. Ця криза триває від 4 днів до 1 тижня. Деякі пацієнти потребують переливання крові, а інші підтримуючого лікування. Гемолітична криза
Гемолітична криза проявляється гострим прискореним падінням рівня гемоглобіну. Червоні кров'яні клітини руйнуються більш швидкими темпами. Це особливо притаманно хворим, у яких є дефіцит глюкозо-6-фосфат-дегідрогенази (G6PD deficiency). При ній використовується підтримуюче лікування, іноді необхідно здійснити переливання крові. Інші види криз
Одним з перших клінічних проявів хвороби є дактиліт, який виникає з 6-місячного віку і може розвиватися у дітей з серповидними ознаками. Тривалість кризи складає місяць. Існує й інший тип серповидної кризи, а саме гострий грудний синдром - це стан, який характеризується лихоманкою, болями в грудях, утрудненим диханням, та інфільтратом, який виявляється за допомогою рентгенівського дослідження грудної клітки. Враховуючи, що пневмонія і серповидні клітини в легенях разом можуть супроводжуватись вище згаданими симптомами, пацієнт проходить лікування від обох. Це може бути викликано болючими кризами, респіраторними інфекціями, емболізацією кісткового мозку або, можливо, ателектазом, лікуванням опіатами або хірургічним втручанням. Ускладнення
Серповидно-клітинна анемія може призвести до різних ускладнень, в тому числі: - нездоланної пост-(авто) спленектомічної інфекції (OPSI), яка пов'язана з функціональною аспленією, яка викликана інкапсульованими організмами, такими як пневмокок (Streptococcus pneumoniae) та гемофільною паличкою (Haemophilus influenzae). Поширеним засобом профілактики виникнення цих захворювань у дитинстві є щоденне вживання пеніциліну (або інших антибіотиків з цієї групи), але деякі лікарі гематологи продовжують курс лікування на невизначений термін. Пацієнти сьогодні отримують користь від планової вакцинації, яка проводиться проти H. Influenzae (гемофілійної палички), S. Pneumoniaе (пневмококу) і Neisseria meningitidis (менінгококу);
- в результаті прогресивного звуження кровоносних судин, що призводить до порушення процесу надходження кисню в мозок, може виникнути інсульт. У дітей зустрічається інфаркт мозку (ішемічний інсульт), а у дорослих трапляються випадки крововиливу в мозок; - жовчнокам'яна хвороба (камені в жовчному міхурі) і холецистит, можуть виникнути в результаті надмірного утворення і випадіння осаду білірубіну спричинений довготривалим гемолізом; - асептичний некроз (асептичний некроз кістки) стегна та ділянок кісток поблизу інших великих суглобів, може бути спричинений ішемією; - зниження імунних реакцій у зв'язку з гіпоспленізмом (hyposplenism) (який має наслідком порушення роботи селезінки); - пріапізм (тривала, зазвичай болюча ерекція, не пов'язана з статевим збудженням) та інфаркт пеніса (перманентна імпотенція);
- остеомієліт (інфекційне захворювання кісткової тканини); найбільш поширеною причиною остеомієліту при серповидно-клітинній анемії є дія сальмонели (Salmonella), (особливо нестандартних серотипів таких як Salmonella Typhimurium, Salmonella Enteritidis, Salmonella choleraesuis і Salmonella paratyphi B), вплив золотистого стафілококу і грамнегативних кишкових бацил призводить до внутрішньосудинного утворення серповидно-клітинних еритроцитів у судинах кишки, що іноді спричиняє ішемічний інфаркт; - толерантність до опіоїдів, може виникнути як нормальна, фізіологічна реакція організму на терапевтичне використання опіатів. Залежність від опіатів зустрічається серед хворих на серповидно-клітинну анемію не частіше ніж серед інших людей, які вживають опіати з інших причин; - гострий папілярний некроз нирок; - трофічні виразки на ногах; - щодо очей, то тут спостерігається фонова ретинопатія, проліферативна ретинопатія, крововилив у скловидне тіло, розшарування сітківки, що в результаті призводить до сліпоти. Саме тому, хворим рекомендується проходити щорічну перевірку очей; -під час вагітності спостерігається затримка внутрішньоутробного розвитку плоду, часті викидні і пре-еклампсії; - хронічний біль. Навіть при відсутності гострого судинно-оклюзійного болю, багато пацієнтів мають хронічні болі, про які вони не повідомляють. - легенева гіпертензія (підвищений тиск у легеневій артерії), призводить до деформації (гіпертрофії та дилатації) правого шлуночка і підвищує ризик виникнення серцевої недостатності; типовими симптомами є задишка, зниження толерантності до фізичного навантаження та напади непритомності, зустрічаються периферичні набряки. - хронічна ниркова недостатність при серповидно-клітинній нефропатії- проявляється гіпертонією (високим кров'яним тиском), протеїнурією (наявність білка в сечі), гематурією (наявність еритроцитів у сечі) і прогресивною анемією. Якщо вона прогресує на кінцевій стадії ниркової недостатності, то прогноз розвитку захворювання у таких хворих дуже негативний. Гетерозиготи Гетерозиготні форми захворювання майже завжди протікають безсимптомно, для них характерні лише прояви ниркової концентрації, які проявляються у вигляді ізостенурії.
Патофізіологія Серповидно-клітинна анемія викликана точковою мутацією ланцюга β-глобіну у гемоглобіні. Ця мутаціяполягає у тому, що у шостій позиції гідрофільні амінокислоти глутамінової кислоти замінюються гідрофобними амінокислотами валіну. Ген β-глобіну знаходиться на короткому плечі 11 хромосоми. Об'єднання двох субодиниць α-глобіну дикого типу з двома мутантними субодиницями β- глобіну призводить до утворення гемоглобіну S (HbS).
У ситуаціях коли вміст кисню у повітрі дуже низький (наприклад на великій висоті), відсутність полярної амінокислоти на шостій позиції у ланцюзі β-глобіну призводить до нековалентної полімеризації (агрегації) гемоглобіну, внаслідок чого змінюється форма червоних кров'яних тілець (вони стають схожими на серп) та суттєво зменшується їх еластичність. Втрата еластичності червоних клітин крові займає центральне місце у патофізіології серповидно-клітинної анемії. Нормальні червоні кров'яні клітини досить еластичні, це дає їм змогу змінювати форму для того, щоб пройти через капіляри. При серповидно-клітинній анемії, недостатність кисню сприяє утворенню серповидно-клітинних еритроцитів, повторення таких процесів призводить до пошкодження клітинної мембрани і зниження еластичності клітин. Тобто, ці клітини не можуть повернутися до своєї нормальної форми навіть після того, як рівень кисню стане нормальним. Як наслідок, новоутворені клітини – дуже жорсткі і не можуть змінювати свою форму, а оскільки вони проходять через вузькі капіляри, то це спричиняє оклюзію судин та ішемію.

Зазвичай, анемія при захворюванні виникає через гемоліз, руйнування еритроцитів у селезінці, через їх неправильну форму. І хоча кістковий мозок утворює нові кров’яні клітини, проте швидкість цього утворення не відповідає швидкості руйнування. Нормальні червоні кров'яні клітини зазвичай живуть 90-120 днів, в той час, як серповидно-клітинні тільки 10-20 днів.
Зазвичай, гемоглобін А складається з двох ланцюгів - альфа-і двох ланцюгів бета-два ланцюги, гемоглобіну А2, складаються з двох альфа-і двох дельта ланцюгів, а гемоглобін F, з двох альфа-і двох гамма ланцюгів. Рівень гемоглобіну в організмі людини на 96-97% складається з гемоглобіну А.
Генетика
Генна мутація, яка викликає серповидно-клітинну анемію, ймовірно виникла спонтанно в різних географічних районах, це припущення ґрунтується на рестрикційному аналізі ендонуклеази. Ці варіації зустрічаються серед населення Камеруну, Сенегалу, Беніну, народів банту та азіатів Саудівської Аравії. Їх клінічне значення полягає в тому, що для деяких з них характерний більш високий рівень HbF, наприклад, при мутації, яка поширена в Сенегалі та Саудівсько-азіатському регіоні, захворювання, як правило, має більш м’яку форму.
У людей, гетерозиготних за HgbS (носії захворювання), проблеми, які виникають через полімеризацію - незначні, адже нормальна алель може виробляти більше 50% гемоглобіну. В осіб, гомозиготних за HgbS, наявність довгих HbS полімерів призводить до спотворення форми еритроцитів. Форма двояко-ввігнутого диска з гладкою поверхнею змінюється на нерівну з великою кількістю шипів, це призводить до того що клітини стають крихкими і тріскають в капілярах.
У носіїв симптоми захворювання з’являються тільки тоді, коли рівень кисню в повітрі дуже знижений (наприклад, на великій висоті) або під час сильного зневоднення організму. Зазвичай, ці кризові ситуації виникають приблизно 0,8 разів на рік в одного пацієнта. Серповидно-клітинна анемія виникає коли сьома амінокислота (якщо рахувати метіонін) - глутамінова кислота замінюється валіном, що спричиняє зміну її структури і функцій.
Це порушення викликане мутацією одного нуклеотиду (відбувається заміна аденіну (А) на тимін (Т)) в гені β-глобіну. Це призводить до того, що у 6 позиції глутамат замінюється валіном. Гемоглобін S, який утворюється в результаті цієї мутації називають HbS, на відміну від нормального гемоглобіну, який має назву HbA. Тобто, досліджуване генетичне захворювання пов’язане з мутацією одного нуклеотиду та, відповідно, зміною послідовності кодону з GAG (ГАГ) на GTG (ГТГ).
Як правило, це доброякісні мутації, які суттєво не впливають на вторинну, третинну, або четвертинну структуру гемоглобіну, при нормальній концентрації кисню. В умовах низької концентрації кисню відбувається процес полімеризації HbS. Деокси-форма гемоглобіну піддається впливу гідрофобної ділянки між Е та F спіралями. Гідрофобні залишки валіну на 6 позиції бета-ланцюга гемоглобіну можуть зв’язуватися з цими гідрофобними ділянками, в результаті чого молекули гемоглобіну S втрачають здатність розчинятися і утворюють так званий волокнистий осад. 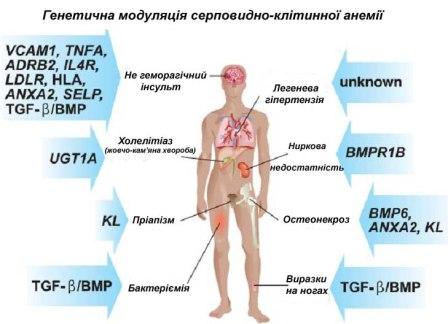 Алель відповідальна за розвиток серповидно-клітинної анемії - аутосомно-рецесивна і розташована на короткому плечі 11 хромосоми. У людини, яка успадковує дефектний ген від батька і матері – розвивається захворювання, тоді як людина, яка отримує одну дефектну копію гена, а одну – нормальну – залишається здоровою, проте є носієм захворювання.
Якщо обидва батьки є носіями серповидно-клітинної анемії, то ймовірність передачі дитині захворювання становить 25%, а ймовірність того, що їх нащадок буде носієм складає – 50%. Але, оскільки відповідальний ген не повністю рецесивний, то в організмі носіїв може утворюватися певна кількість серповидно-клітинних еритроцитів, проте їхня кількість не на стільки велика, щоб викликати симптоми хвороби, проте достатня для того щоб хворі були стійкими до дії малярії. Саме тому у гетерозиготних осіб рівень пристосування – вищий ніж у гомозиготних. Це явище відоме під назвою гетерозиготна перевага.
Саме через цю перевагу, хвороба, як і раніше дуже поширена, особливо часто вона зустрічається серед населення у зв'язку з адаптивною перевагою гетерозигот, особливо серед людей предки яких походять із тих територій, де малярія була дуже поширена, зокрема, Африка, Середземномор'я, Індія та Близький Схід. Раніше, епідемії малярії часто спостерігалися на території Південної Європи, проте це захворювання було повністю ліквідоване тут у середині ХХ ст. і зараз не зустрічається за винятком рідкісних спорадичних випадків.
Збудник малярії має складний життєвий цикл, частина якого проходить в еритроцитах (червоних кров’яних тільцях). У носіїв серповидно-клітинної анемії, наявність малярійних паразитів призводить до того, що змінені кров’яні тільця (з дефектним типом гемоглобіну) розриваються передчасно, внаслідок чого плазмодій (збудник малярії) не може відтворити ці клітини.
Крім того, полімеризація гемоглобіну впливає в першу чергу на здатність паразита перетравлювати Hb. Таким чином, у тих районах, де поширена малярія, шанси людей на виживання насправді збільшуються, якщо вони є носіями серповидно-клітинної хвороби.
У США, де малярія майже не зустрічається, поширеність серповидно-клітинної анемії серед чорношкірого населення нижча (близько 0,25%), ніж у Західній Африці (близько 4,0%) і, варто зазначити, цей рівень постійно зменшується. Якщо не враховувати позитивний вплив серповидно-клітинної анемії на населення країн Африки, то мутація, яка викликає це захворювання абсолютно – не корисна для людської популяції, саме тому, вони швидше за все в результаті селективного відбору зникне. Іншим фактором, що обмежує поширення серповидно-клітинних генів в Північній Америці є відсутність культурної схильності до полігамії (характерна для ісламських країн).
Успадкування СКА успадковується від батьків за такими ж принципами, як і група крові, колір і структура волосся, колір очей та інші фізичні риси. Тип гемоглобіну людини у червоних клітинах крові залежить від того, які гени гемоглобіну були успадковані від батьків. Якщо один з батьків має серповидно-клітинну анемію (SS), а інший має серповидно-клітинну особливість, тобто є лише носієм хвороби (АS),то в такому випадку існує 50% вірогідність успадкування дитиною як хвороби так і ознаки. Якщо обоє батьків мають серповидно-клітинну ознаку (АS), то дитина з ймовірністю 25% (1 з 4) буде хворою (SS). Детальнішу інформацію про серповидно-клітинну анемію Ви можете знайти у статті
<<<
Алькаптонурія
Гістидинемія
Гомоцистинурія
Полікістозна хвороба нирок
Хвороба Вільсона – Коновалова
Cиндром Марфана (Хвороба Марфана)
X-зчеплений іхтіоз
Анемія Фанконі
Аргініносукцинатна ацидурія
Бета-таласемія
Бічний аміотрофічний склероз (частина друга)
Бічний аміотрофічний склероз (частина перша)
Галактоземія
Гемоглобін Е
Гемоглобін С
Гемохроматоз
Глікогенози
Дальтонізм (частина друга)
Дальтонізм (частина перша)
Дефіцит 3-гідкрокси-метил-глутарил КоА ліази
Дефіцит 3-метилкротоніл-коензим А карбоксилази
Дефіцит альфа 1 антитриптисину
Дефіцит бета-кетотіолази
Дефіцит біотинідази
Дефіцит дигідропіримідин дегідрогенази
Дефіцит довголанцюгової ацил-коензим А дегідрогенази
Дефіцит коротколанцюгової ацил-коензим А дегідрогенази
Дефіцит метилентетрагідрофолат редуктази
Дефіцит прекалікреїну
Дефіцит синтетази голокарбоксилази
Дефіцит фактора ХІ
Ентеропатичний акродерматит
Іміногліцинурія
М'язова дистрофія Дюшена
Муковісцидоз
Нейрофіброматоз
Органічні ацидемії
Первинний системний дефіцит карнітину
Перманентний неонатальний цукровий діабет
Перонеальна дистрофія Шарко-Марі -Тута (частина друга)
Перонеальна дистрофія Шарко-Марі-Тута (частина перша)
Пропіонова ацидурія
Псевдополідистрофія Гурлера
Серповидно-клітинна анемія (частина друга)
Серповидно-клітинна анемія (частина перша)
Синдром Ангельмана
Синдром Бартера
Синдром Блума
Синдром Дабіна Джонсона
Синдром Дауна (частина друга)
Синдром Дауна (частина перша)
Синдром Едвардса
Синдром Клайнфельтера-Рейфенштейна-Олбрайта
Синдром котячого крику (5р)
Синдром Кріглера Найара
Синдром Леша-Наяна
Синдром Лойса-Дітца
Синдром Патау
Синдром Прадера-Віллі
Сімейна вегетативна дисфункція
Сімейна гіперхолестеринемія
Тирозинемія
Тирозинемія І типу
Тирозинемія ІI типу
Тирозинемія ІIІ типу
Фенілкетонурія (ФКУ)
Фруктоземія
Хвороба I клітин
Хвороба Гірке
Хвороба Дауна
Хвороба Канавана
Хвороба кленового сиропу
Хвороба Краббе
Хвороба Німана – Піка
Хвороба Тея-Сакса (друга частина)
Хвороба Тея-Сакса (перша частина)
Хвороба Фабрі
Хвороба фон Віллебранда
Хвороба Хантінгтона
Целіакія (частина друга)
Целіакія (частина перша)
Церебротендінальний ксантоматоз
|