


Генетичні захворюванняСиндром Прадера-Віллі Синдром Прадера-Віллі (скорочено СПВ) - це рідкісне генетичне захворювання, при якому сім генів (або деякі їх частини) на 15 батьківській хромосомі (Q 11-13) – видалені або нормально не функціонують (наприклад при частковій делеції хромосоми 15Q). Вперше розлад був описаний у 1956 році Андреа Прадером і Генріхом Віллі, Алексіс Лабхарт, Ендрю Зіглером і Гвідо Фанконі. СПВ зустрічається в 1 особи на 25000-10000 новонароджених. Дуже важливо пам’ятати, що генетичний матеріал, який впливає на розвиток захворювання - батьківський. Тому що для цієї області 15 хромосоми характерним є явище імпринтингу. А це означає, що у деяких генів цього регіону лише одна копія гену функціонує нормально, через імпринтинг.
У генах, які спричиняють розвиток СПВ копія гена, що була отримана від батька – функціонує, в той час, як материнська – ні. Це означає, що в той час як більшість людей мають одну робочу копію цих генів, люди з синдромом Прадера-Віллі не мають цієї копії. На сьогодні відомо один розлад, який називають «сестринською» хворобою синдрому Прадера Віллі – це синдром Ангельмана, при якому мутації піддається материнський генетичний матеріал аналогічного генетичного регіону. Ознаки і симптоми захворювання
Клінічні особливості і ознаки Холм та ін. (1993) описують ті симптоми та ознаки, наявність яких дає змогу поставити попередній діагноз – синдром Прадера-Віллі, навіть якщо вони будуть присутні не всі: Внутрішньоутробні ознаки:
* зниження активності руху плоду; * ненормальне положення плоду; * багатоводдя (надмірна кількість амніотичної рідини). Ознаки при народженні: * сідничне передлежання плоду або народження з допомогою кесаревого розтину; * летаргія; * гіпотонія; * труднощі при годуванні (через поганий м'язовий тонус, який впливає на смоктальний рефлекс); * труднощі при диханні; * гіпогонадизм. Ознаки в ранньому дитинстві: * затримка фізичного розвитку (продовжуються труднощі при годуванні); 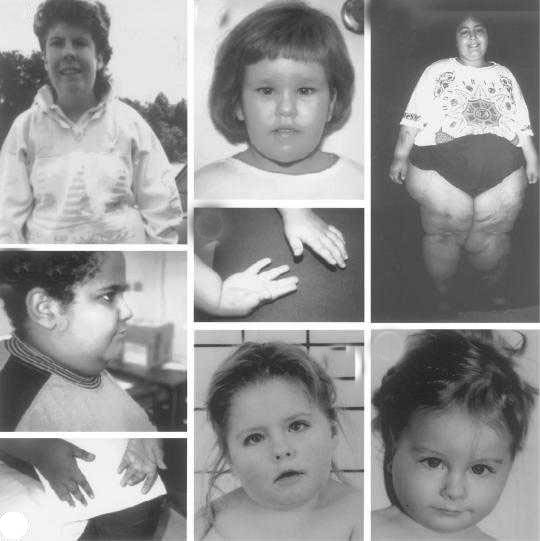 * затримка інтелектуального розвитку; * швидка втомлюваність (надмірна сонливість); * косоокість; * сколіоз (часто не виявляється при народженні). Ознаки в дитинстві: * затримка розвитку мовленнєвих навичок; * погана фізична координація; * гіперфагія (переїдання) у віці від 2 - 8 років; * надмірне збільшення ваги;
* порушення сну; * сколіоз. Ознаки в підлітковому віці: * затримка статевого дозрівання; * низький зріст; * ожиріння; * аномальна гнучкість. Ознаки у повнолітті: * безпліддя (чоловіки та жінки); * гіпогонадизм; * рідке лобкове волосся; * ожиріння; * гіпотонія; * труднощі при навчанні / обмежені інтелектуальні функції (але в деяких випадках рівень інтелекту може бути середній); * підвищена схильність до розвитку цукрового діабету; * аномальна гнучкість. Загальні зовнішні ознаки (для дорослих): * великий та широкий ніс; * маленькі руки та ноги з вузенькими пальцями; * чутлива шкіра (легко з’являються синці); * надлишкові жирові відкладення, особливо в центральній частині тіла; * високий, вузький лоб; * мигдалеподібні очі з тонкими, опущеними вниз повіками; * шкіра і волосся світліше ніж усіх інших членів сім’ї; * порушення нормального статевого розвитку. * дерматіломанія (skin picking);
* поява розтяжок (стрій); * затримка моторного розвитку. Нейро-когнітивні відхилення У осіб з СПВ виникають труднощі при навчанні та при концентрації уваги. Курф і Фрим (Curfs and Frym) (1992) вивчали різноманітні ступені розумових відхилень (та труднощів, які виникають при навчанні) серед осіб, хворих на СПВ. Результати їхнього дослідження були наступними: * 5%: хворих мають IQ вище 85 (низький середній рівень інтелекту); * 27%: IQ 70 - 85 (межа інтелектуальної діяльності); * 39%: IQ 50 - 70 (незначна розумова відсталість); * 27%: IQ 35 - 50 (помірна розумова відсталість); * 1%: IQ 20 - 35 (важка розумова відсталість); * <1%: IQ <20 (глибока розумова відсталість). Кессіді (Cassidy) виявив, що 40% осіб із синдромом Прадера-Віллі мають рівень інтелекту дещо нижче середнього або знаходиться на межі інтелектуальних здібностей, як бачимо, ця цифра дещо нижча за дані Курфа і Фрима (Curfs and Frym) (32%). Проте, більшість досліджень показує, що переважна кількість людей (50-65 %) це ті, хто мають незначну розумову відсталість - їхній інтелектуальний рівень можна назвати перехідним та ті рівень інтелектуального розвитку яких дещо нижчий середнього. Діти з СПВ мають дещо незвичний когнітивний профіль. У них часто дуже добре розвинута візуальне сприйняття, в т. ч. вони добре читають і мають багатий словниковий запас, проте їхні мовленнєві здібності (які іноді порушуються через гіперназальність (порушення голосу)), суттєво поступаються їхньому розумінню. Слухова і послідовна обробка інформації знаходиться на досить низькому рівні, так само як здібності до математичних дисциплін і письма. У хворих погіршена зорова та слухова короткострокова пам'ять і звукова концентрація уваги. Іноді, рівень розвитку з віком покращується, проте дефіцит інтелектуальних здібностей в цих областях все одно спостерігається. Поведінкові розлади
Синдром Прадера-Віллі часто пов'язаний з аномальним підвищенням апетиту, що часто призводить до патологічного ожиріння. На сьогодні не існує єдиної думки щодо причин появи цього синдрому, хоча генетичні порушення в 15 хромосомі можуть порушити нормальне функціонування гіпоталамуса. З огляду на те, що гіпоталамус регулює багато основних процесів, в тому числі і апетит, його пошкодження може бути цілком реальною причиною появи вище зазначеного симптому. Проте при патологоанатомічному дослідженні жодних морфологічних змін у структурі гіпоталамуса, які б могли викликати таке порушення виявлено не було. Особи, уражені синдромом Прадера-Віллі мають підвищений рівень греліну в організмі. Як вважають вчені, саме ця речовина відіграє безпосередню роль у підвищенні апетиту, гіперфагії та подальшому ожирінні. Кессіді вважав, що необхідно чіткого розмежувати поведінкові прогнози та відновлення інтелектуальних здібностей і створити окремий комплекс регулярних процедур, для кожної групи порушень. Основні психічні розлади, які виникають у хворих на СПВ – це компульсивна поведінка, (яка зазвичай проявляється у вигляді посмикування за шкіру (skin-picking) і підвищеної тривожності), психіатричні симптоми, наприклад, такі як галюцинації, параноя і депресія, які можуть виникати приблизно в 5-10% хворих молодих людей. Психіатричні та поведінкові проблеми є найчастішою причиною госпіталізації хворих на СПВ. Ендокринні порушення Є кілька аспектів СПВ, які дають змогу підтвердити концепцію дефіциту гормону росту в осіб з синдромом Прадера-Віллі. По перше, хворі люди, зазвичай мають низький ріст, страждають ожирінням при аномальній будові тіла, тобто у них знижений вміст вільної жирової маси, знижений рівень LBM і зменшений загальний обсяг витраченої енергії та знижену щільність кісткової тканини. Для СПВ, характерною рисою є гіпогонадизм. Він проявляється у вигляді неопущення яєчок у чоловіків і появи доброякісної передчасної адренархе у жінок. Сім’яники можуть опуститися з часом, або цей недолік можна виправити хірургічною операцією або здійсненням замісної терапії тестостероном. Адренархе можна лікувати використовуючи методи замісної гормональної терапії. Генетика
СПВ викликається делецією батьківської копії імпринтованих SNRPN гену малого ядерного рибонуклепротеїнового поліпептиду N і гену necdin, який знаходиться поряд з кластерами м-РНК: SNORD64, SNORD107, SNORD108 і двома копіями SNORD109, 29 копією SNORD116 (HBII-85) і 48 копією SNORD115 (HBII -52). Вони розташовані на 15 хромосомі в регіоні 15q11-13. Це так званий PWS/AS регіон може бути втрачений в результаті дії одного із кількох генетичних механізмів в більшості випадків через мутації. Інші менш поширені механізми включають: унібатьківську дисомію (uniparental disomy), випадкові мутації, хромосомні транслокації та делеції гена. Через дію імпринтингу, активність копій вище зазначених генів, які були успадковані від матері дуже низька (або взагалі відсутня), тобто виражені тільки батьківські копії генів. СПВ виникає в результаті втрати активності батьківської копії цього регіону. Якщо делеції відбуваються у цьому ж регіоні на материнській хромосомі, то це призводить до виникнення синдрому Ангельмана. Синдром Прадера-Віллі та синдром Ангельмана – це перші описані випадки порушення процесу імпринтингу людини.
Ризик народження хворої дитини у сім’ї, де вже є один хворий нащадок, залежить від генетичного механізму, який викликав розлад. Ймовірність народження ураженої дитини становить менше 1%, якщо у неї спостерігається делеція гена або однобатьківська дисомія, якщо ж у дитини мутація регіону, для якого характерне явище імпринтингу, то ця ймовірність зростає до 50%, у випадку появи хромосомних транслокацій, ризик виникнення розладу в наступної дитини становить 25%. Для діагностики всіх відомих механізмів можливе використання пренатального тестування. Дослідження, які проводяться за участю груп людей та, ті які здійснювалися на моделях мишей, показали, що видалення 29 копії C/D box snoRNA SNORD116 (HBII-85) є основною причиною виникнення синдрому Прадера-Віллі. Діагностика
СПВ виникає приблизно в 1 особи з 10000-25000 новонароджених. У всьому світі на сьогодні є понад 400000 людей, які живуть з СПВ. Як вже було сказано, це захворювання традиційно характеризується гіпотонією, невеликим зростом, гіперфагією, ожирінняя, поведінковими проблемами. В осіб з цим розладом маленькі руки і ноги, для них характерний гіпогонадизм і легка розумова відсталість. Проте, якщо діагностувати дане захворювання на ранньому етапі і розпочати його лікування доступними засобами, то прогноз розвитку захворювання стає більш оптимістичним. СПВ, так само як аутизм – це захворювання, яке має дуже широкий спектр проявів і ознак. Перебіг хвороби відрізняється в кожному окремому випадку і може варіюватися від легкої форми до важкої протягом всього життя людини. Синдром Прадера-Віллі впливає на різні органи і системи. Зазвичай, діагноз синдром Прадера-Віллі ставиться на основі клінічних проявів. Проте сьогодні все частіше використовується генетичне тестування, яке особливо рекомендується для новонароджених з гіпотонією. Рання діагностика дозволяє здійснювати раннє лікування СПВ. Для дітей з синдромом рекомендуються щоденні ін’єкції рекомбінантного гормону росту (GH). Соматотропін (соматотропний гормон гіпофізу) підтримує постійне збільшення м'язової маси і може зменшити апетит хворого. Основою діагностики розладу, як вже було сказано, є генетичне тестування, яке може проводитися методом ДНК-метилювання, для виявлення того, чи на хромосомі 15q11-q13 присутній нормально функціонуючий регіон, відхилення у якому призводять до появи синдромів Прадера-Віллі та Ангельмана. Така перевірка дозволяє виявити більш ніж 97% пацієнтів. Таке тестування необхідно здійснювати для того, щоб підтвердити діагноз СПВ, особливо у новонароджених (адже вони ще дуже маленькі, щоб можна було перевірити їхні здібності, які дають змогу діагностувати хворобу за клінічними проявами). Оскільки при народженні немовлят з синдромом Прадера-Віллі виникають деякі труднощі, то варто пам’ятати, що вроджені травми і кисневе голодування можуть ускладнити генетичні недоліки, в результаті атипового СПВ.
Диференційна діагностика
Часто, синдром Прадера-Віллі неправильно діагностується. Причиною цього є те, що багато лікарів не знають про цей синдром. Іноді його вважають синдромом Дауна, через те, що цей розлад зустрічається набагато частіше ніж СПВ. Крім того, характерне для СПВ ожиріння, може бути присутнім також при синдромі Дауна через поведінкові проблеми. Проблем додає і той факт, що батьки дітей, які вже здійснювали тестування для діагностики синдрому Прадера-Віллі можуть розповісти друзям, родині і навіть лікарям і медичним сестрам, що їх дитина має синдром Дауна, тому що про цей розлад знають більше людей. Вважається, що близько 75% випадків СПВ залишаються невиявленими. Лікування
Для лікування СПВ на сьогодні немає жодних ефективних ліків. Декілька препаратів, спрямованих на подолання симптомів захворювання зараз знаходяться на стадії розробки. У дитинстві, хворі особи повинні пройти лікування, яке б допомогло покращити тонус м'язів. Дуже важливою є фізіотерапія. Протягом навчального року, хворі діти повинні отримувати додаткову допомогу, а процес навчання повинен бути дуже гнучким. Найбільшою проблемою, яка пов'язана із СПВ є серйозне ожиріння. Через важке ожиріння, поширеним ускладненням є обструктивне апное сну, саме тому, часто необхідним може бути використання Сiпап (індивідуального медичного апарату для автоматизованої тривалої допоміжної інтраназальної вентиляції легень постійним позитивним тиском). Суспільство і культура Вперше публічна інформація про синдром Прадера-Віллі з'явилася у британських ЗМІ в липні 2007 року, коли телевізійний канал Channel 4 показав програму під назвою Can't Stop Eating («Не можу припинити їсти»), в якій описувалося щоденне життя двох осіб з СПВ - Джо і Тамари. Актриса і невролог Маїма Бялик написала дисертацію про синдром Прадера-Віллі для отримання кандидатського ступеня у 2008 році. <<<
Алькаптонурія
Гістидинемія
Гомоцистинурія
Полікістозна хвороба нирок
Хвороба Вільсона – Коновалова
Cиндром Марфана (Хвороба Марфана)
X-зчеплений іхтіоз
Анемія Фанконі
Аргініносукцинатна ацидурія
Бета-таласемія
Бічний аміотрофічний склероз (частина друга)
Бічний аміотрофічний склероз (частина перша)
Галактоземія
Гемоглобін Е
Гемоглобін С
Гемохроматоз
Глікогенози
Дальтонізм (частина друга)
Дальтонізм (частина перша)
Дефіцит 3-гідкрокси-метил-глутарил КоА ліази
Дефіцит 3-метилкротоніл-коензим А карбоксилази
Дефіцит альфа 1 антитриптисину
Дефіцит бета-кетотіолази
Дефіцит біотинідази
Дефіцит дигідропіримідин дегідрогенази
Дефіцит довголанцюгової ацил-коензим А дегідрогенази
Дефіцит коротколанцюгової ацил-коензим А дегідрогенази
Дефіцит метилентетрагідрофолат редуктази
Дефіцит прекалікреїну
Дефіцит синтетази голокарбоксилази
Дефіцит фактора ХІ
Ентеропатичний акродерматит
Іміногліцинурія
М'язова дистрофія Дюшена
Муковісцидоз
Нейрофіброматоз
Органічні ацидемії
Первинний системний дефіцит карнітину
Перманентний неонатальний цукровий діабет
Перонеальна дистрофія Шарко-Марі -Тута (частина друга)
Перонеальна дистрофія Шарко-Марі-Тута (частина перша)
Пропіонова ацидурія
Псевдополідистрофія Гурлера
Серповидно-клітинна анемія (частина друга)
Серповидно-клітинна анемія (частина перша)
Синдром Ангельмана
Синдром Бартера
Синдром Блума
Синдром Дабіна Джонсона
Синдром Дауна (частина друга)
Синдром Дауна (частина перша)
Синдром Едвардса
Синдром Клайнфельтера-Рейфенштейна-Олбрайта
Синдром котячого крику (5р)
Синдром Кріглера Найара
Синдром Леша-Наяна
Синдром Лойса-Дітца
Синдром Патау
Синдром Прадера-Віллі
Сімейна вегетативна дисфункція
Сімейна гіперхолестеринемія
Тирозинемія
Тирозинемія І типу
Тирозинемія ІI типу
Тирозинемія ІIІ типу
Фенілкетонурія (ФКУ)
Фруктоземія
Хвороба I клітин
Хвороба Гірке
Хвороба Дауна
Хвороба Канавана
Хвороба кленового сиропу
Хвороба Краббе
Хвороба Німана – Піка
Хвороба Тея-Сакса (друга частина)
Хвороба Тея-Сакса (перша частина)
Хвороба Фабрі
Хвороба фон Віллебранда
Хвороба Хантінгтона
Целіакія (частина друга)
Целіакія (частина перша)
Церебротендінальний ксантоматоз
|