


Генетичні захворювання : Аутосомно-домінантні хворобиНейрофіброматоз Нейрофіброматоз (скорочено NF; нейрофіброматоз 1 типу також відомий як хвороба фон Реклінгхаузена) - це генетичне спадкове захворювання, при якому з нервової тканини утворюються пухлини (нейрофіброми), які можуть бути як доброякісними так і можуть завдати серйозної шкоди організму через стиснення нервів та інших тканин. Розлад впливає на всі клітини нервового гребеня (клітини Шванна (шванівські клітини, лемоцити), меланоцити і ендоневральні фібробласти). Клітинні елементи цих типів клітин надмірно розмножуються по всьому організму, утворюючи пухлини. При цьому захворюванні, меланоцити також функціонують неправильно, в результаті чого з’являються безпричинні пігментні плями та інші прояви порушеної пігментації. Пухлини можуть викликати появу підшкірних гуль, з’являються кольорові плями, скелетні проблеми, виникає тиск на основу спинномозкових нервів та інші неврологічні проблеми.
Нейрофіброматоз - аутосомно-домінантне захворювання, тобто якщо особа успадкує навіть одну копію дефектного гена, то у неї цей розлад все одно буде розвиватися. Так, якщо тільки один із батьків має нейрофіброматоз, то його (її) діти мають 50% вірогідність розвитку розладу. Важкість проявів захворювання, може змінюватися, в залежності від рівня експресії гена. Приблизно половина випадків захворювання виникає через спонтанні мутації, тобто в осіб без сімейно історії захворювання. Кількість хворих на нейрофіброматоз чоловіків і жінок приблизно однакова.
Класифікація - Нейрофіброматоз 1 типу (NF 1) - також відомий як "хвороба фон Реклінгхаузена" є найбільш поширеною формою NF, на яку припадає близько 90% випадків.
- Нейрофіброматоз 2 типу (також відомий як "центральний нейрофіброматоз") – виникає в результаті мутації гену merlin (інша назва якого "schwannomin" або NР2), Ознаки і симптоми
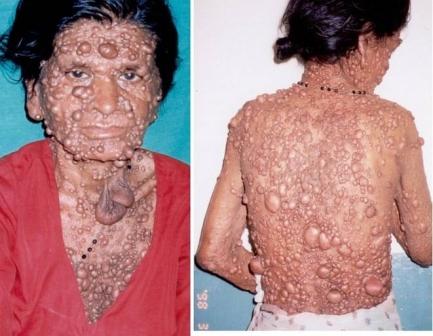 Нейрофіброматоз по різному проявляється у хворих осіб. Розлад дуже часто проявляється у виглядіплексиформних нейром, оптичних гліом або неврином слухового нерва, але випадки смертності пацієнтів найчастіше пов'язані зі злоякісною трансформацією нейром таких, як нейрофібросаркоми (така трансформація дуже рідко (менш ніж у 3% випадків) відбувається у хворих з NF1). Нейрофіброматоз по різному проявляється у хворих осіб. Розлад дуже часто проявляється у виглядіплексиформних нейром, оптичних гліом або неврином слухового нерва, але випадки смертності пацієнтів найчастіше пов'язані зі злоякісною трансформацією нейром таких, як нейрофібросаркоми (така трансформація дуже рідко (менш ніж у 3% випадків) відбувається у хворих з NF1).
Існує високий рівень ризику появи різноманітних розумових відхилень та когнітивних дефектів, особливо характерними такі порушення є для осіб з NF-1, проте ознак серйозної розумової відсталості серед пацієнтів, зазвичай не спостерігається. Через прогресивний розвиток пухлин, які впливають на діяльність нервової системи, а також у зв’язку з розвитком макроцефалії (у дітей) при нейрофіброматозі існує підвищений ризик виникнення епілепсії.
Крім того, нейрофіброматоз збільшує ризик розвитку лейкемії, переважно у дітей. Так, у дітей з NF-1 ризик появи цього захворювання у 200-500 разів більший ніж у загальної популяції. Оскільки характерні для захворювання пухлини утворюються, за участю нервових клітин, то вони можуть виникати у видимих частинах тіла, що може призводити до появи у хворих суттєвих соціальних проблем. Трапляються випадки, коли пухлини розвиваються в таких областях, де вони можуть призводити до появи інших медичних проблем і, відповідно для безпеки пацієнта, ці злоякісні утворення необхідно видалити.
Для цього, може бути потрібним проведення кількох хірургічних операцій. Це може бути обмежуючі втручання або операції, які проводяться з допомогою гамма-ножа, в залежності від того, де ці пухлини розташовані. Наприклад, особам з NF 2 для запобігання розвитку глухоти, варто провести хірургічну декомпресію пухлин ділянки преддверно-равликового нерва.
Пов’язані розлади
Нейрофіброматоз відноситься до групи захворювань, відомих під назвою факоматози (phakomatoses, neurocutaneous syndromes ). Окрім типів нейрофіброматозу, факоматози також включають туберозний склероз, синдром Стурге-Вебера (Sturge-Weber syndrome) і хворобу фон Хіппеля-Ліндау (von Hippel-Lindau disease). Ці захворювання віднесли до однієї групи – раніше, ще до того часу, коли стала зрозумілою генетична природа порушення.
Генетика
NF-1 і NF-2 може бути успадкований за аутосомно-домінантним типом, або ж виникати в результаті випадкових мутацій (мутації de novo). Нейрофіброматоз 1 типу пов'язаний з мутацією на хромосомі 17q11.2. Цей ген кодує діяльність нейрофібріміну (фермент, який активує ГТФ-азу (GAP)). Нейрофіброматоз 2 типу пов'язаний з мутацією в хромосомі 22q, генним продуктом є merlin – цитоскелетний білок.
Як NF-1 так і NF-2 – це аутосомно-домінантні розлади. Тобто, для того, щоб дитина була хворою на нейрофіброматоз, їй достатньо успадкувати лише одну копію дефектного гена від когось із батьків. Якщо у дитини один із батьків хворий на NF-1 або NF-2, а інший – здоровий, то ризик успадкування захворювання становить 50% -100% в залежності від того, чи той з батьків, котрий хворий є гомозиготним (АА) за цією ознакою чи гетерозиготним (Аа) ("А" означає дефектну домінантну алель, "а" - рецесивну алель).
Питання спадковості ускладнюється також через відмінності між генотипом і фенотипом, тобто типовими генетичними і фактичними проявами хвороби. При захворюванні на нейрофіброматоз 1 типу, суттєвого зв’язку між генотипом і фенотипом поки що не виявлено. Важкість і специфіка розладу може дуже відрізнятися у кожного хворого члена сім’ї. Це яскравий приклад явища зі змінною експресивністю - коли перебіг захворювання відрізняється у людей з однаковим генотипом.
Проте, у разі захворювання на NF-2, прояви розладу у членів сім’ї дуже схожі. Тобто, як вважається між генотипом та фенотипом при цьому типі фіброматозу існує сильний зв'язок. Ймовірно, що NF-1 і NF-2 можуть виникати через появу спонтанної мутації, тобто не передаються по спадковості. Таку природу мають близько 50% випадків захворювання на нейрофіброматоз.
Так само, як полідактилія, нейрофіброматоз теж є аутосомно-домінантним захворюванням, яке серед населення зустрічається рідко. На нейрофіброматоз-1 типу хворіє приблизно 1 особа з 2500-3000 новонароджених (частота носіїв - 0,0004, генна частота 0,0002) і цей тип більш поширений ніж NF-2.
Патофізіологія
Дефектний ген, який відповідає за виникнення нейрофіброматозу 1 типу розташований на довгому плечі хромосоми 17 (q11.2). Він кодує білок, нейрофібромін та пригнічує діяльність онкобілка p21. При нейрофіброматозі функціональність цього білка змінюється або послаблюється (через делеції, міссенс мутації, або нонсенс-мутації). Внаслідок цього, кількість клітин у всьому тілі збільшується (в т.ч. навколо нервової системи).
Основною проблемою при захворюванні є неможливість інактивації GTP через дефектну ГТФ-азу (нейрофібромін). Це призводить до появи характерних для нейрофіброматозу симптомів появи великих пухлин (нейрофібром і шваном). Щодо гену, мутації в якому викликають NF-2,то на сьогодні, інформації про нього значно менше, але відомо, що він має назву merlin, розташований на довгому плечі хромосоми 22q (11.1-13.1) і кодує діяльність білка з аналогічною назвою.
Лікування
Оскільки жодних ефективних ліків для лікування захворювання немає, то для уражених осіб можливе лише здійснення професійного симптоматичного підтримуючого лікування для подолання різноманітних симптомів та ускладнень. При стисненні органів пухлинами необхідним може бути хірургічне втручання. У 10 % випадків захворювання в уражених осіб можуть розвиватися ракові новоутворення. У цих випадках доцільним є використання хіміотерапії.
Хоча на сьогодні немає жодного відомого методу лікування NF, проте члени Асоціації з питань нейрофіброматозу (Neurofibromatosis Association) оптимістично налаштовані на те, що в найближчі 5-10 років, буде знайдено нове ефективне лікування для подолання цього розладу. Для сімей, де були випадки нейрофіброматозу, доступний генетичний скринінг та генетична консультація.
Історія
Нейрофіброматоз (або хвороба фон Реклінгхаузена) був вперше описаний в 1882 році німецьким патологоанатом, Фрідріхом Даніелем фон Реклінгхаузеном. У студентські роки Реклінгхаузен був учнем відомого вченого Рудольфа Вірхова, який працював у Берліні. Саме Реклінгхаузен здійснив одні з найбільш важливих медичних спостережень того часу. Він був першим хто описав і запропонував термін для характеристики такого захворювання як гемохроматоз. Сьогодні Реклінгхаузен відомий за його внесок у розробку методів забарвлення і, головним його досягненням стала його робота про нейрофіброматоз, опублікована у 1881 році.
Суспільство і культура
Відомі випадки
У листопаді 2006 року в Великобританії був представлений документальний фільм про організацію Facing the World, яка допомагає дітям з важкими недоліками зовнішності у країнах що розвиваються. Одним із дітей, про яких йшла мова у фільмі був індонезійський хлопчик Аріанто, який був уражений важкою формою нейрофіброми, яка проявлялася гігантичним збільшенням половини обличчя.
Крім того, в цьому ж році, з’явився інший документальний фільм BBC2 (який мав назву «Грані життя»), де теж описувався випадок розвитку нейрофіброматозу. У цьому документальному фільмі розповідали про підлітка Аміта Гхоша, який у 14 років вирішив що йому необхідно проведення коректуючої операції для виправлення певних дефектів зовнішності, спричинених хворобою. У нього на обличчі з’явилася нейрофіброма, в результаті розвитку якої він втратив зір на одне око. А для того, щоб видалити цю пухлину необхідно було видалити й око. Це був випадок NF-2, при якому після суттєвого пошкодження однієї сторони обличчя, інша сторона залишилася абсолютно нормальною.
У січні 2008 року, 32-річному китайцю Хуангу Чункаю (Huang Chuncai) провели другу операцію з видалення 4,5 кг (9.9 фунтів) пухлини з його обличчя. Під час першого хірургічного втручання було видалено 15 кг (33 фунтів) пухлини, загальна маса якої спочатку становила 23 кг (55,7 фунта).
У березні 2007 року, лікування 30-річного француза Паскаля Колера, який був хворий на нейрофіброматоз, закінчилося після того як йому здійснили першу в світі операцію з повної трансплантації обличчя.
Раніше вважалося, що Джозеф Меррік (якого ще називають Людина-слон), уражений нейрофіброматозом 1 типу. Проте, як виявилося, він хворий дуже рідкісним синдромом Протея. Проте ця ситуація, породила поширену помилку, що нейрофіброматоз і хвороба "людини-слона" – це одне і те ж захворювання. На сьогодні існує декілька національних організацій, які надають підтримку людям, хворим на нейрофіброматоз. Основні з них такі:
* Дитяча пухлинна фундація (The Children's Tumor Foundation (CTF)) - некомерційна організація, діяльність якої спрямована на лікування нейрофіброматозу в Сполучених Штатах Америки);
* Корпорація Нейрофіброматоз (Neurofibromatosis Inc) - некомерційна організація, яка здійснює підтримку осіб, хворих на нейрофіброматоз у США);
* Кафе нейрофіброматоз (Neurofibromatosis Cafe) - некомерційна організація, яка надає хворій на нейрофіброматоз особі всі можливі знання про перебіг захворювання, методи лікування, тощо, дія на території США;
* Фундація «Маленька жаба» (Little Frog Foundation) – це некомерційна організація, діяльність якої спрямована на надання всієї можливої інформації про NF1, NF2 хворим та членам їх сімей, діє на території Австралії.
* NF KONTAKT.be - некомерційна організація, що надає інформаційні ресурси про працівників освітніх та медичних закладів, які займаються проблемами NF1, NF2, і пухлино-зв'язаного нейрофіброматозу в Бельгії і забезпечує обізнаність і підтримку хворим на нейрофіброматоз у Європі.
<<<
Алькаптонурія
Гістидинемія
Гомоцистинурія
Полікістозна хвороба нирок
Хвороба Вільсона – Коновалова
Cиндром Марфана (Хвороба Марфана)
X-зчеплений іхтіоз
Анемія Фанконі
Аргініносукцинатна ацидурія
Бета-таласемія
Бічний аміотрофічний склероз (частина друга)
Бічний аміотрофічний склероз (частина перша)
Галактоземія
Гемоглобін Е
Гемоглобін С
Гемохроматоз
Глікогенози
Дальтонізм (частина друга)
Дальтонізм (частина перша)
Дефіцит 3-гідкрокси-метил-глутарил КоА ліази
Дефіцит 3-метилкротоніл-коензим А карбоксилази
Дефіцит альфа 1 антитриптисину
Дефіцит бета-кетотіолази
Дефіцит біотинідази
Дефіцит дигідропіримідин дегідрогенази
Дефіцит довголанцюгової ацил-коензим А дегідрогенази
Дефіцит коротколанцюгової ацил-коензим А дегідрогенази
Дефіцит метилентетрагідрофолат редуктази
Дефіцит прекалікреїну
Дефіцит синтетази голокарбоксилази
Дефіцит фактора ХІ
Ентеропатичний акродерматит
Іміногліцинурія
М'язова дистрофія Дюшена
Муковісцидоз
Нейрофіброматоз
Органічні ацидемії
Первинний системний дефіцит карнітину
Перманентний неонатальний цукровий діабет
Перонеальна дистрофія Шарко-Марі -Тута (частина друга)
Перонеальна дистрофія Шарко-Марі-Тута (частина перша)
Пропіонова ацидурія
Псевдополідистрофія Гурлера
Серповидно-клітинна анемія (частина друга)
Серповидно-клітинна анемія (частина перша)
Синдром Ангельмана
Синдром Бартера
Синдром Блума
Синдром Дабіна Джонсона
Синдром Дауна (частина друга)
Синдром Дауна (частина перша)
Синдром Едвардса
Синдром Клайнфельтера-Рейфенштейна-Олбрайта
Синдром котячого крику (5р)
Синдром Кріглера Найара
Синдром Леша-Наяна
Синдром Лойса-Дітца
Синдром Патау
Синдром Прадера-Віллі
Сімейна вегетативна дисфункція
Сімейна гіперхолестеринемія
Тирозинемія
Тирозинемія І типу
Тирозинемія ІI типу
Тирозинемія ІIІ типу
Фенілкетонурія (ФКУ)
Фруктоземія
Хвороба I клітин
Хвороба Гірке
Хвороба Дауна
Хвороба Канавана
Хвороба кленового сиропу
Хвороба Краббе
Хвороба Німана – Піка
Хвороба Тея-Сакса (друга частина)
Хвороба Тея-Сакса (перша частина)
Хвороба Фабрі
Хвороба фон Віллебранда
Хвороба Хантінгтона
Целіакія (частина друга)
Целіакія (частина перша)
Церебротендінальний ксантоматоз
|