


Генетичні захворювання : Полігенні хворобиПеронеальна дистрофія Шарко-Марі-Тута (частина перша) Хвороба Шарко-Марі-Тута (ШМТ), відома також як невральна аміотрофія Шарко-Марі, спадкова сенсомоторна нейропатія - є спадковим захворюванням нервів (невропатія), яке приймає різні форми. Вона характеризується втратою м’язової тканини та сенсорного відчуття, переважно у ступнях та ногах, але це явище (на пізніх стадіях захворювання) характерне також для рук та плечей. На даний момент захворювання є невиліковним. ШМТ є одним з найбільш поширених спадкових неврологічних розладів, який зустрічається у 36 осіб із 100000.
Молекулярні причини виникнення
Хвороба Шарко-Марі-Тута спричинена мутаціями, які викликають дефекти у білках нейронів. Нервові сигнали проводяться аксонами, які вкриті мієлінової оболонкою. Більшість мутацій при ШМТ уражають мієлінову оболонку та деякі аксони.
Найбільш поширеною причиною виникнення хвороби (70-80% випадків) є дублювання великого регіону в хромосомі 17p12, що включає в себе ген РМР22. Деякі мутації уражають MFN2 ген, який кодує діяльність мітохондріального білка. Клітини містять окремі набори генів в їх ядрі та мітохондріях. У нервових клітинах, мітохондрії рухаються донизу вздовж довгого аксону. При певних формах ШМТ, мутований MFN2 ген спричиняє утворення мітохондрією великого кластера, або згустка, який не може рухатися вниз по аксону до синапсів, що в свою чергу порушує їх функціональність.
Виділяють такі типи хвороби: первинна демієлінізуюча нейропатія (ШМТ1, ШМТ3 і ШМТ4) та первинна аксональна нейропатія (ШМТ2), з частими випадками накладання цих типів між собою. Інші клітини які впливають на появу хвороби - це лемоцити (Шванівські клітини), які створюють мієлінову оболонку, шляхом обгортання плазматичної мембрани навколо аксонів, ця структура, іноді порівнюється зі швейцарським рулетом.
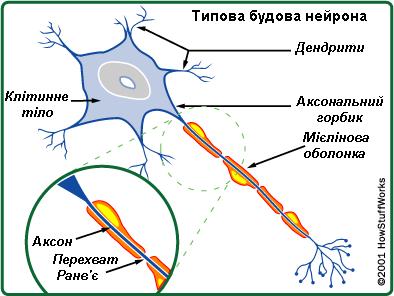 Нейрони, шванівські клітини та фібробласти працюючи спільно утворюють здоровий (працюючий) нерв. Шванівські клітини і нейрони проводять молекулярні сигнали, які регулюють численні процеси в організмі. Саме ці сигнали порушені при хворобі Шарко-Марі-Тута.
Демієлінізація шванівських клітин викликає порушення структури і функцій аксонів. Які, можуть стати причиною дегенерації або порушення функціональності аксона. Мієлінова оболонка дозволяє нервовим клітинам проводити сигнали значно швидше. Якщо мієлінова оболонка пошкоджується, то швидкість проведення нервових сигналів уповільнюються. Швидкість сигналів може бути визначена шляхом проведення електроміографії – дуже поширеного неврологічного дослідження. Також, коли аксон є пошкодженим це призводить до зменшення біопотенціалу м’язів (CMAP).
Симптоми
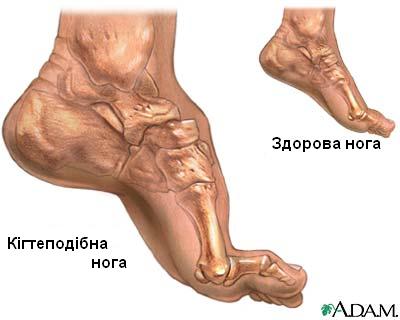
Симптоми ШМТ, зазвичай, починають проявлятись у пізньому дитинстві або ранньому дорослому віці. Деякі люди не відчувають жодних ознак до того часу поки їм не виповняється тридцять або сорок років. Як правило, первинними симптомами захворювання є труднощі при тильному згинанні ноги та гомілки. Це може також викликати появу кігтеподібної ноги, коли пальці на ногах мають аномальну, скручену форму. Атрофія м'язової тканини нижньої частини ніг спричиняє деформацію ноги, що призводить до появи так званої "ноги лелеки" або "перевернутої пляшки". З прогресуванням хвороби у багатьох людей у більш пізньому віці з’являється слабкість в руках і передпліччях.
 Симптоми і перебіг хвороби може варіювати. У деяких випадках порушується дихання крім того ураженим може бути слух, зір, шия та плечові м'язи. Поширеним явищем є сколіоз. Не виключеним є пошкодження кульшової западини. При хворобі можливі шлунково-кишкові розлади, труднощі при жуванні, ковтанні і мовленні (атрофія медіального краю голосової складки). Атрофія м’язів може викликати тремор. Як правило, вагітність загострює ШMT, так само як сильне емоційне напруження.
Нейропатичний біль часто є симптомом ШMT, хоча, як й інші симптоми його наявність і тяжкість змінюється в кожному окремому випадку. У деяких людей, біль може бути дуже сильним і заважати у повсякденному житті. Проте не у всіх уражених хвороба супроводжується болем. Коли біль присутня як симптом ШMT, її характер такий самий як при інших периферичних нейропатіях, таких як постгерпетична невралгія та комплексний регіональний больовий синдром, та ін.
Діагностика
Хвороба Шарко-Марі-Тута може бути діагностована через наявність характерних симптомів та за допомогою вимірювання швидкості електроміографії, біопсії нерва, а також шляхом аналізу ДНК. ДНК-тестування може дати остаточний, достовірний діагноз, але не всі генетичні маркери для ШMT на сьогодні є відомими. Першим симптомом хвороби є слабкість у гомілці та деформація стопи, така як важкість при тильному згинанні ноги та щиколотки, молотоподібні пальці та високий підйом. Але самі по собі ознаки не є підставою для встановлення діагнозу хвороби, саме тому пацієнти повинні бути направлені до невролога або реабілітолога (фізіотерапевта).
.JPG) Щоб оцінити м’язову слабкість невролог попросить пацієнта пройти на носочках або перемістити частину ноги проти сили опору. З метою виявлення рівня чутливості невролог буде визначати глибокі сухожильні рефлекси (ті, які визначаються з допомогою молоточка), а саме колінний рефлекс (який при хворобі знижений або відсутній). Лікар також запитає про сімейну історію захворювання через спадковий характер передачі ШМТ. Відсутність сімейного анамнезу не виключає наявність хвороби Шарко-Марі-Тута, але це дозволяє лікареві виключити інші причини невропатії, такі як діабет або вплив певних хімічних речовин чи наркотиків. Щоб оцінити м’язову слабкість невролог попросить пацієнта пройти на носочках або перемістити частину ноги проти сили опору. З метою виявлення рівня чутливості невролог буде визначати глибокі сухожильні рефлекси (ті, які визначаються з допомогою молоточка), а саме колінний рефлекс (який при хворобі знижений або відсутній). Лікар також запитає про сімейну історію захворювання через спадковий характер передачі ШМТ. Відсутність сімейного анамнезу не виключає наявність хвороби Шарко-Марі-Тута, але це дозволяє лікареві виключити інші причини невропатії, такі як діабет або вплив певних хімічних речовин чи наркотиків. У 2010 році, хвороба ШMT була одним з перших захворювань, для якого за допомогою секвенування генома ураженої людини була точно визначена генетична причина хвороби. У гені були виявлені дві мутації, з яких саме мутація SH3TC2 була названа причиною виникнення хвороби. Потім дослідники порівняли геном пацієнта з геномами матері, батька та семи братів і сестер хворого з і без хвороби. Мати і батько мали одну нормальну та одну мутовану копії цього гена, у зв’язку з чим симптоми були хвороби були помірними, або їх взагалі не було. У потомства, що успадкувало дві копії аномальних генів хвороба проявлялась у повному обсязі. Початкова вартість секвенування генома пацієнта складала близько 50 тисяч дол., але дослідники підрахували, що незабаром воно коштуватиме менше $ 5000 і стане загально доступним.
Типи
Станом на початок 2010 року, було ідентифіковано мутації у 39 генах, які спричиняють появу ШMT. Хворобу спочатку можна класифікувати за основними клінічними категоріями, а потім за підтипами у відповідності з цими мутаціями. Тип 1, в першу чергу, впливає на мієлінову оболонку і є або домінуючим або рецесивним Х-зчепленим. 2 тип першочергово впливає на аксон і є або домінуючим або рецесивним. Інші типи є змішаними. Клінічні категорії
Детальнішу інформацію про хворобу Шарко-Марі-Тута Ви зможете знайти у статті: "Перонеальна дистрофія Шарко-Марі-Тута (частина друга)" <<<
Алькаптонурія
Гістидинемія
Гомоцистинурія
Полікістозна хвороба нирок
Хвороба Вільсона – Коновалова
Cиндром Марфана (Хвороба Марфана)
X-зчеплений іхтіоз
Анемія Фанконі
Аргініносукцинатна ацидурія
Бета-таласемія
Бічний аміотрофічний склероз (частина друга)
Бічний аміотрофічний склероз (частина перша)
Галактоземія
Гемоглобін Е
Гемоглобін С
Гемохроматоз
Глікогенози
Дальтонізм (частина друга)
Дальтонізм (частина перша)
Дефіцит 3-гідкрокси-метил-глутарил КоА ліази
Дефіцит 3-метилкротоніл-коензим А карбоксилази
Дефіцит альфа 1 антитриптисину
Дефіцит бета-кетотіолази
Дефіцит біотинідази
Дефіцит дигідропіримідин дегідрогенази
Дефіцит довголанцюгової ацил-коензим А дегідрогенази
Дефіцит коротколанцюгової ацил-коензим А дегідрогенази
Дефіцит метилентетрагідрофолат редуктази
Дефіцит прекалікреїну
Дефіцит синтетази голокарбоксилази
Дефіцит фактора ХІ
Ентеропатичний акродерматит
Іміногліцинурія
М'язова дистрофія Дюшена
Муковісцидоз
Нейрофіброматоз
Органічні ацидемії
Первинний системний дефіцит карнітину
Перманентний неонатальний цукровий діабет
Перонеальна дистрофія Шарко-Марі -Тута (частина друга)
Перонеальна дистрофія Шарко-Марі-Тута (частина перша)
Пропіонова ацидурія
Псевдополідистрофія Гурлера
Серповидно-клітинна анемія (частина друга)
Серповидно-клітинна анемія (частина перша)
Синдром Ангельмана
Синдром Бартера
Синдром Блума
Синдром Дабіна Джонсона
Синдром Дауна (частина друга)
Синдром Дауна (частина перша)
Синдром Едвардса
Синдром Клайнфельтера-Рейфенштейна-Олбрайта
Синдром котячого крику (5р)
Синдром Кріглера Найара
Синдром Леша-Наяна
Синдром Лойса-Дітца
Синдром Патау
Синдром Прадера-Віллі
Сімейна вегетативна дисфункція
Сімейна гіперхолестеринемія
Тирозинемія
Тирозинемія І типу
Тирозинемія ІI типу
Тирозинемія ІIІ типу
Фенілкетонурія (ФКУ)
Фруктоземія
Хвороба I клітин
Хвороба Гірке
Хвороба Дауна
Хвороба Канавана
Хвороба кленового сиропу
Хвороба Краббе
Хвороба Німана – Піка
Хвороба Тея-Сакса (друга частина)
Хвороба Тея-Сакса (перша частина)
Хвороба Фабрі
Хвороба фон Віллебранда
Хвороба Хантінгтона
Целіакія (частина друга)
Целіакія (частина перша)
Церебротендінальний ксантоматоз
|