


Генетические заболевания : Аутосомно-рецессивные болезниСиндром Блума Синдром Блума (СБ), (англ. Bloom syndrome), также известен как синдром Блума-Торре-Махачека - это редкое аутосомно-рецессивное хромосомное расстройство, которое характеризуется высокой частотой разрывов и перестроек в хромосомах больного человека. Этот синдром был впервые описан врачом дерматологом Девидом Блумом (Dr. David Bloom) в 1954 году. Признаки и симптомы.
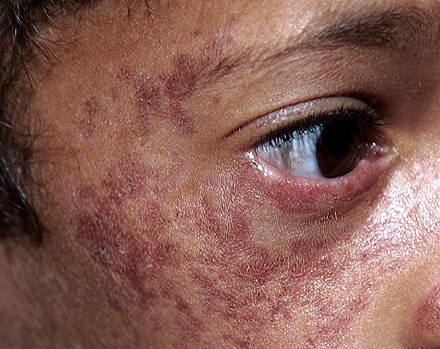 Лица, больные ВБ, как правило не высокого роста, с характерными высыпаниями на коже, которыевозникают почти сразу после первого воздействия солнечных лучей. Эти высыпания, как правило возникают на щеках (вызывая покраснение) и имеют форму бабочки. Но сыпь можетт возникать не только на лице, но и на других частях тела, на которые попадали солнечные лучи (спина, руки и т.д.). Другие клинические признаки:
Осложнения, возникающие в результате воздействия заболевания, могут включать в себя хронические болезни легких, сахарный диабет, возникновение трудностей при обучении. У некоторых лиц, возникает умственная отсталость. Наиболее опасным осложнением можно назвать повышение вероятности возникновения опухолевых заболеваний.
Связь с возникновением рака  Вследствие большого количества мутаций, происходящих при заболевании синдромом Блума, у больныхсущественно повышается риск заболеть раком. Предрасположенность к онкологическим заболеваниям характеризуется: 1) широким спектром проявления болезней, включающий лейкозы, лимфомы и карциномы; 2) ранним возрастом возникновения опухолевых заболеваний, поровну к остальному населению; 3) сложностью и тяжестью течения.
Рис. 2. Микрогнатия
У лиц с синдромом Блума рак может возникнуть в любом возрасте. Средний возраст больных - 25 лет. Патофизиология Мутации в гене BLM, который является составной ДНК геликаз (хеликазы), вызывают возникновение синдрома Блума. ДНК геликазы (хеликазы) - это ферменты, которые раскручивают двухцепную спираль ДНК. Процесс «раскрутки» состоит из большого количества процессов, таких как синтез копий ДНК, транскрипция РНК, репарация ДНК и т.д. Когда клетка готовится к делению, то хромосомы дублируются таким образом, что каждая новообразованная структура получает полный набор хромосом. Процесс удвоения называется - репликацией ДНК. Ошибки, допущенные при репликации ДНК могут привести к мутации. Белок BLM играет важную роль в поддержании стабильности ДНК в процессе репликации. Мутации гена BLM, связанные с синдромом Блума дезактивируют белок, который кодируется геном BLM и входит в состав ДНК хеликазы, или же нарушают процесс экспрессии (образование белка). Другими словами не происходит экспрессия этого гена. Отсутствие BLM белка или низкая его активность приводит к увеличению количества мутаций, именно поэтому молекулярные механизмы, с помощью которых BLM поддерживает стабильность хромосом, до сих пор остаются предметом для многих исследований.
У лиц, с СБ имеет место большое количество обменов между гомологичными хромосомами или сестринскими хроматидами (две молекулы ДНК, образующиеся в процессе репликации ДНК), в результате которых существенно увеличивается количество мутаций. Кроме того, на много чаще (в отношении лиц, не имеющих синдрома Блума) случается повреждение хромосом и их перестройка. Сегодня ученые работают над тем, что установить (или опровергнуть) наличие прямой связи между молекулярными процессами, в которых участвует ген BLM и изменением хромосом. Кроме того, связь между молекулярными дефектами в клетках лиц с СБ, хромосомными мутациями, которые накапливаются в соматических клетках (всех клетках тела, кроме половых) и многими клиническими признаками синдрома Блума также является одним из направлений клинических исследований. Синдром Блума наследуется по аутосомно-рецессивному типу. То есть, оба родителя должны быть носителями для того, чтобы их ребенок заболел. Носителями заболевания среди евреев Ашкенази, преимущественно являются выходцами из Восточной Европы, является 1 человек на 100. Если оба родителя являются носителями СБ, то в 25% случаев, ребенок может быть больным. Тем членам семей, которые могут быть носителями заболевания рекомендуется обратиться с просьбой о генетической консультации или пройти генетическое тестирование. Для носителей-родителей, которые планируют рожать ребенка следует провести пренатальную диагностику с помощью цитогенетических и молекулярных методов. Также возможно осуществить молекулярный анализ ДНК для определения наличия мутаций. Другие мутации при синдроме Блума Впервые две миссенс мутации фермента пируват киназы М2 (H391Y и K422R), обнаружили в клетках больных синдромом Блума, как потом подтвердилось они повышают предрасположенность к возникновению рака. Результаты показывают, что несмотря на наличие мутаций в области домена мижсубединичного контакта, мутантные белки K422R и H391Y сохраняют свою гомотетраметрическую структуру, что делает их похожими по структуре на обычные белки, но их активность существенно уменьшается на 75 и 20% соответственно. Интересно, что H391Y показал 6-кратное увеличение родства со своим субстратом фосфоэнолпируватом и вел себя как неалостерический белок с нарушенным соединением (с субстратом). Однако, K422R теряет свое родство с фосфоэнолпируват. В отличие от K422R, H391Y проявляет улучшенные тепловые характеристики и стабильность в определенных диапазонах значений рН, кроме того наблюдается меньшее влияние алостеричних ингибиторов Phe, и устойчивость во время структурных изменений при связывании активатора (фруктозо 1,6-бисфосфата) и ингибитора (Phe ). Оба мутанты показали небольшой сдвиг в оптимуме рН от 7,4 до 7,0.Совместная экспрессия гомотетрамеров дикого типа и мутанта PKM2 в клеточной среде, которая возникает в результате взаимодействия между ними на уровне мономеров, была позднее подтверждена в ходе клинических экспериментов. Кросс-мономерное взаимодействие существенно изменяет олигомерное состояние PKM2, через усиление димеризации и гетеротетрамеризации Эти особенности, как предполагается, играют важную роль при опухолевой прогрессии. PKM2 (M2-PK); потенциал многофункционального белка. Высказано предположение, что аберрантный белок PKM2 может быть основной причиной возникновения других симптомов, характерных для синдрома Блума. <<<
Алькаптонурия
Гистидинемия
Гомоцистинурия
Поликистозная болезнь почек
Cиндром Марфана (Болезнь Марфана)
Анемия Фанкони
Аргининосукцинатная ацидурия
Бета-талассемия
Боковой амиотрофический склероз (часть вторая)
Боковой амиотрофический склероз (часть первая)
Болезнь I клеток
Болезнь Вильсона - Коновалова
Болезнь Гирке
Болезнь Дауна
Болезнь Канавана
Болезнь кленового сиропа
Болезнь Краббе
Болезнь Ниманна - Пика
Болезнь Тея-Сакса (вторая часть)
Болезнь Тея-Сакса (первая часть)
Болезнь Фабри
Болезнь фон Виллебранда
Болезнь Хантингтона
Галактоземия
Гемоглобин Е
Гемоглобин С
Гемохроматоз
Гликогенозы
Дальтонизм (часть вторая)
Дальтонизм (часть первая)
Дефицит 3 гидрокси-метил-глутарил-КоА лиазы
Дефицит 3-метилкротонил-коэнзим А карбоксилазы
Дефицит альфа 1 антитриптисина
Дефицит бета-кетотиолазы
Дефицит биотинидазы
Дефицит дигидропиримидин дегидрогеназы
Дефицит длинноцепочечной ацил-коэнзим А дегидрогеназы
Дефицит короткоцепочечной ацил-коэнзим А дегидрогеназы
Дефицит метилентетрагидрофолат редуктазы
Дефицит прекалликреин
Дефицит синтетазы голокарбоксилазы
Дефицит фактора ХI
Иминоглицинурия
Муковисцидоз
Мышечная дистрофия Дюшенна
Нейрофиброматоз
Органические ацидемии
Первичный системный дефицит карнитина
Перманентный неонатальный сахарный диабет
Перонеальная дистрофия Шарко-Мари-Тута (часть вторая)
Перонеальная дистрофия Шарко-Мари-Тута (часть первая)
Пропионовая ацидурия
Псевдополидистрофия Гурлера
Семейная вегетативная дисфункция
Семейная гиперхолестеринемия
Серповидно-клеточная анемия (часть вторая)
Серповидно-клеточная анемия (часть первая)
Синдром Ангельмана
Синдром Бартера
Синдром Блума
Синдром Дабина Джонсона
Синдром Дауна (часть вторая)
Синдром Дауна (часть первая)
Синдром Клайнфельтера-Рейфенштейна-Олбрайта
Синдром кошачьего крика
Синдром Криглера Найара
Синдром Леша-Наяна
Синдром Лойса-Дитца
Синдром Патау
Синдром Прадера-Вилли
Синдром Эдвардса
Тирозинемия
Тирозинемия І типа
Тирозинемия ІI типа
Тирозинемия ІIІ типа
Фенилкетонурия (ФКУ)
Фруктоземия
Х-сцепленный-ихтиоз
Целиакия (часть вторая)
Целиакия (часть первая)
Церебротендинальный ксантоматоз
Цистинурия
|