


Генетические заболеванияСиндром Прадера-Вилли Синдром Прадера-Вилли (сокращенно СПВ) - это редкое генетическое заболевание, при котором семь генов (или некоторые их части) на 15 отцовской хромосоме (Q 11-13) - удалены или нормально не функционируют (например, при частичной делеции хромосомы 15Q). Впервые расстройство было описано в 1956 году Андреа Прадером и Генрихом Вилли, Алексис Лабхарт, Эндрю Зиглером и Гвидо Фанкони. СПВ встречается у 1 человека на 25000-10000 новорожденных. Очень важно помнить, что генетический материал, который влияет на развитие заболевания - отцовский. Потому что для этой области 15 хромосомы характерно явление импринтинга. А это значит, что у некоторых генов этого региона только одна копия гена функционирует нормально, через импринтинг.
В генах, вызывающих развитие СПВ копия гена, которая была получена от отца - функционирует, в то время как материнская - нет. Это означает, что в то время как большинство людей имеют одну рабочую копию этих генов, люди с синдромом Прадера-Вилли не имеют этой копии. На сегодня известно одно заболевание, которое называют «сестринской» болезнью синдрома Прадера Вилли - это синдром Ангельмана, при котором мутации подвергается материнский генетический материал аналогичного генетического региона. Признаки и симптомы заболевания
Клинические особенности и признаки
Холм и др. (1993) описывают те симптомы и признаки, наличие которых позволяет поставить предварительный диагноз - синдром Прадера-Вилли, даже если они будут присутствовать не все:
Внутриутробные признаки: * Снижение активности движения плода;
* Ненормальное положение плода;
* Многоводие (чрезмерное количество амниотической жидкости).
Признаки при рождении:
* Ягодичное предлежание плода или рождения с помощью кесарева сечения;
* Летаргия;
* Гипотония;
* Трудности при кормлении (из-за плохого мышечного тонуса, который влияет на сосательный рефлекс);
* Трудности при дыхании;
* Гипогонадизм.
Признаки в раннем детстве:
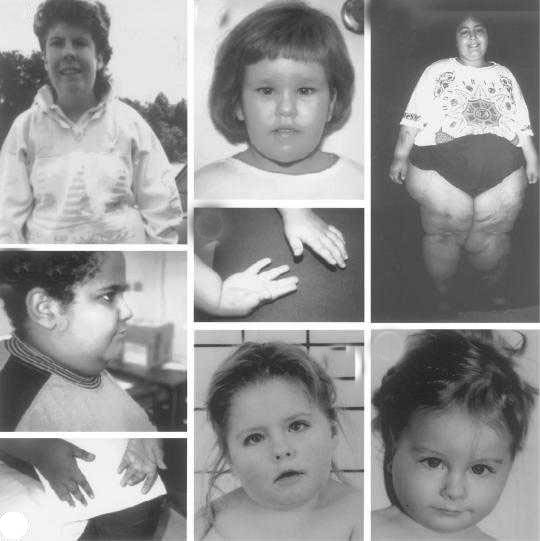 * Задержка физического развития (продолжаются трудности при кормлении);
* Задержка интеллектуального развития;
* Быстрая утомляемость (чрезмерная сонливость);
* Косоглазие;
* Сколиоз (часто не проявляется при рождении).
Признаки в детстве:
* Задержка развития речевых навыков;
* Плохая физическая координация;
* Гиперфагия (переедание) в возрасте от 2 - 8 лет;
* Чрезмерное увеличение веса;
* Нарушение сна;
* Сколиоз.
Признаки в подростковом возрасте:
* Задержка полового созревания;
* Низкий рост;
* Ожирение;
* Аномальная гибкость.
Признаки при совершеннолетии:
* Бесплодие (мужчины и женщины);
* Гипогонадизм;
* Жидкие лобковые волосы;
* Ожирение;
* Гипотония;
* Трудности при обучении / ограниченные интеллектуальные функции (но в некоторых случаях уровень интеллекта может быть средний);
* Повышенная склонность к развитию сахарного диабета;
* Аномальная гибкость.
Общие внешние признаки (для взрослых):
* Большой и широкий нос;
* Маленькие руки и ноги с узенькими пальцами;
* Чувствительная кожа (легко появляются синяки);
* Избыточные жировые отложения, особенно в центральной части тела;
* Высокий, узкий лоб;
* Миндалевидные глаза с тонкими, опущенными веками;
* Кожа и волосы светлее, чем у всех других членов семьи;
* Нарушение нормального полового развития.
* Дерматиломания (skin picking);
* Появление растяжек;
* Задержка моторного развития.
Нейро-когнитивные отклонения
У людей с СПВ возникают трудности при обучении и при концентрации внимания. Курф и Фрим (Curfs and Frym) (1992) изучали различные степени умственных отклонений (и трудностей, возникающих при обучении) среди лиц, больных СПВ. Результаты их исследования были следующими:
* 5%: больных имеют IQ выше 85 (низкий средний уровень интеллекта);
* 27%: IQ 70 - 85 (граница интеллектуальной деятельности);
* 39%: IQ 50 - 70 (незначительная умственная отсталость);
* 27%: IQ 35 - 50 (умеренная умственная отсталость);
* 1%: IQ 20 - 35 (тяжелая умственная отсталость);
* <1%: IQ <20 (глубокая умственная отсталость).
Кессиди (Cassidy) обнаружил, что 40% лиц с синдромом Прадера-Вилли имеют уровень интеллекта несколько ниже среднего или находится на грани интеллектуальных способностей, как видим, эта цифра несколько ниже данных Курфа и Фрима (Curfs and Frym) (32%). Однако большинство исследований показывает, что подавляющее количество людей (50-65%) это те, кто имеют незначительную умственную отсталость - их интеллектуальный уровень можно назвать переходным и те уровень интеллектуального развития которых несколько ниже среднего.
Дети с СПВ имеют несколько необычный когнитивный профиль. У них часто хорошо развито визуальное восприятие, в т.ч. они хорошо читают и имеют богатый словарный запас, однако их речевые способности (которые иногда нарушаются из-за гиперназальности (нарушение голоса)), существенно уступают их пониманию.
Слуховая и последовательная обработка информации находится на достаточно низком уровне, равно как способности к математическим дисциплинам и письму. У больных ухудшена зрительная и слуховая краткосрочная память и звуковая концентрация внимания. Иногда, уровень развития с возрастом улучшается, однако дефицит интеллектуальных способностей в этих областях все равно наблюдается.
Поведенческие расстройства Синдром Прадера-Вилли часто связан с аномальным повышением аппетита и часто приводит к патологическому ожирению. На сегодня не существует единого мнения относительно причин возникновения этого синдрома, хотя генетические нарушения в 15 хромосоме могут нарушить нормальное функционирование гипоталамуса. Ввиду того, что гипоталамус регулирует многие основные процессы, в том числе и аппетит, его повреждение может быть вполне реальной причиной появления вышеуказанного симптома. Однако при патологоанатомическом исследовании никаких морфологических изменений в структуре гипоталамуса, которые могли бы вызвать такое нарушение, обнаружено не было.
Лица, пораженные синдромом Прадера-Вилли, имеют повышенный уровень грелина в организме. Как считают ученые, именно это вещество играет непосредственную роль в повышении аппетита, гиперфагии и дальнейшем ожирении. Кессиди считал, что необходимо четко разграничить поведенческие прогнозы и восстановление интеллектуальных способностей и создать отдельный комплекс регулярных процедур, для каждой группы нарушений.
Основные психические расстройства, возникающие у больных СПВ - это компульсивное поведение, (которое обычно проявляется в виде подергивания за кожу (skin-picking) и повышенной тревожности), психиатрические симптомы, например, такие как галлюцинации, паранойя и депрессия, которые могут возникать примерно в 5-10% больных молодых людей. Психиатрические и поведенческие проблемы являются наиболее частой причиной госпитализации больных с СПВ.
Эндокринные нарушения
Есть несколько аспектов СПВ, позволяющих подтвердить концепцию дефицита гормона роста у лиц с синдромом Прадера-Вилли. Во первых, больные люди, обычно имеют низкий рост и страдают ожирением при аномальном строении тела, т.е. у них пониженное содержание свободной жировой массы, пониженный уровень LBM и уменьшен общий объем израсходованной энергии и пониженная плотность костной ткани.
Для СПВ, характерной чертой является гипогонадизм. Он проявляется в виде неопущения яичек у мужчин и появления доброкачественной преждевременной адренархе у женщин. Семенники могут опуститься со временем, или этот недостаток можно исправить хирургической операцией или осуществлением заместительной терапии тестостероном. Адренархе можно лечить, используя методы заместительной гормональной терапии.
Генетика
СПВ вызывается делецией родительской копии импринтированных SNRPN гена малого ядерного рибонуклепротеинового полипептида N и гена necdin, который находится рядом с кластерами мРНК: SNORD64, SNORD107, SNORD108 и двумя копиями SNORD109, 29 копией SNORD116 (HBII-85) и 48 копией SNORD115 (HBII -52). Они расположены на 15 хромосоме в регионе 15q11-13. Это так называемый PWS/AS регион может быть потерян в результате действия одного из нескольких генетических механизмов в большинстве случаев через мутации. Другие менее распространенные механизмы включают: униотцовскую дисомию (uniparental disomy), случайные мутации, хромосомные транслокации и делеции гена.
Из-за действия импринтинга, активность копий вышеуказанных генов, которые были унаследованы от матери очень низкая (или вообще отсутствует), то есть выражены только родительские копии генов. СПВ возникает в результате потери активности родительской копии этого региона. Если делеции происходят в этом же регионе на материнской хромосоме, то это приводит к возникновению синдрома Ангельмана. Синдром Прадера-Вилли и синдром Ангельмана - это первые описанные случаи нарушения процесса импринтинга человека.
Риск рождения больного ребенка в семье, где уже есть один больной потомок, зависит от генетического механизма, который вызвал расстройство. Вероятность рождения больного ребенка составляет менее 1%, если у него наблюдается делеция гена или униотцовская дисомия, если же у ребенка мутация региона, для которого характерно явление импринтинга, то эта вероятность возрастает до 50%, в случае появления хромосомных транслокаций, риск возникновения болезни у следующего ребенка составляет 25%. Для диагностики всех известных механизмов, возможно использование пренатального тестирования.
Исследования, проводимые с участием групп людей и те, которые осуществлялись на моделях мышей, показали, что удаление 29 копий C/D box snoRNA SNORD116 (HBII-85) является основной причиной возникновения синдрома Прадера-Вилли.
Диагностика
СПВ возникает примерно у 1 человека из 10000-25000 новорожденных. Во всем мире на сегодня есть более 400000 человек, которые живут с СПВ. Как уже было сказано, это заболевание традиционно характеризуется гипотонией, небольшим ростом, гиперфагией, ожирением, поведенческими проблемами. У лиц с этим расстройством маленькие руки и ноги, для них характерен гипогонадизм и легкая умственная отсталость.
Однако если диагностировать данное заболевание на раннем этапе и начать его лечение, то прогноз развития заболевания становится более оптимистичным. СПВ, равно как аутизм - это заболевание, которое имеет очень широкий спектр проявлений и признаков. Течение болезни отличается в каждом отдельном случае и может варьироваться от легкой формы до тяжелой, которая прогрессирует в течение всей жизни человека. Синдром Прадера-Вилли влияет на различные органы и системы. Обычно диагноз синдром Прадера-Вилли ставится на основании клинических проявлений. Однако сегодня все чаще используется генетическое тестирование, которое особенно рекомендуется для новорожденных с гипотонией. Ранняя диагностика позволяет осуществлять раннее лечение СПВ. Для детей с синдромом рекомендуются ежедневные инъекции рекомбинантного гормона роста (GH). Соматотропин (соматотропный гормон гипофиза) поддерживает постоянное увеличение мышечной массы и может уменьшить аппетит больного.
Основой диагностики расстройства, как уже было сказано, является генетическое тестирование, которое может проводиться методом ДНК-метилирования, для выявления того, присутствует ли на хромосоме 15q11-q13 нормально функционирующий регион, отклонения в котором приводят к появлению синдромов Прадера-Вилли и Ангельмана. Такая проверка позволяет выявить более 97% пациентов. Такое тестирование необходимо осуществлять для того, чтобы подтвердить диагноз СПВ, особенно у новорожденных (ведь они еще очень маленькие, чтобы можно было проверить их способности, позволяющие диагностировать болезнь по клиническим проявлениям).
Поскольку при рождении младенцев с синдромом Прадера-Вилли возникают некоторые трудности, то следует помнить, что врожденные травмы и кислородное голодание могут осложнить генетические недостатки, в результате атипичного СПВ.
Дифференциальная диагностика
Часто, синдром Прадера-Вилли неправильно диагностируется. Причиной этого является то, что многие врачи не знают об этом синдроме. Иногда его считают синдромом Дауна, потому что это расстройство встречается гораздо чаще СПВ. Кроме того, характерное для СПВ ожирения, может присутствовать также при синдроме Дауна через поведенческие проблемы.
Проблем добавляет и тот факт, что родители детей, которые уже осуществляли тестирования для диагностики синдрома Прадера-Вилли, могут рассказать друзьям, семье и даже врачам и медицинским сестрам, что их ребенок имеет синдром Дауна, потому что об этом расстройстве знают больше людей. Считается, что около 75% СПВ остаются не выявленными. Лечение
Для лечения СПВ на сегодня нет никаких эффективных лекарств. Ряд препаратов, направленных на преодоление симптомов заболевания сейчас находятся на стадии разработки. В детстве, больные лица должны пройти лечение, которое бы помогло улучшить тонус мышц. Очень важна физиотерапия. В течение учебного года, больные дети должны получать дополнительную помощь, а процесс обучения должен быть очень гибким. Наибольшей проблемой, связанной с СПВ есть серьезное ожирение.
Из-за тяжелого ожирения, распространенным осложнением является обструктивное апноэ сна, именно поэтому, часто необходимым может быть использование СИПАП (индивидуального медицинского аппарата для автоматизированной длительной вспомогательной интраназальной вентиляции легких постоянным положительным давлением).
Общество и культура
Впервые публичная информация о синдроме Прадера-Вилли появилась в британских СМИ в июле 2007 года, когда телевизионный канал Channel 4 показал программу под названием Can't Stop Eating («Не могу прекратить есть»), в которой описывалось ежедневную жизнь двух человек из СПВ - Джо и Тамары.
Актриса и невролог Маима Бялик написала диссертацию о синдроме Прадера-Вилли для получения кандидатской степени в 2008 году <<<
Алькаптонурия
Гистидинемия
Гомоцистинурия
Поликистозная болезнь почек
Cиндром Марфана (Болезнь Марфана)
Анемия Фанкони
Аргининосукцинатная ацидурия
Бета-талассемия
Боковой амиотрофический склероз (часть вторая)
Боковой амиотрофический склероз (часть первая)
Болезнь I клеток
Болезнь Вильсона - Коновалова
Болезнь Гирке
Болезнь Дауна
Болезнь Канавана
Болезнь кленового сиропа
Болезнь Краббе
Болезнь Ниманна - Пика
Болезнь Тея-Сакса (вторая часть)
Болезнь Тея-Сакса (первая часть)
Болезнь Фабри
Болезнь фон Виллебранда
Болезнь Хантингтона
Галактоземия
Гемоглобин Е
Гемоглобин С
Гемохроматоз
Гликогенозы
Дальтонизм (часть вторая)
Дальтонизм (часть первая)
Дефицит 3 гидрокси-метил-глутарил-КоА лиазы
Дефицит 3-метилкротонил-коэнзим А карбоксилазы
Дефицит альфа 1 антитриптисина
Дефицит бета-кетотиолазы
Дефицит биотинидазы
Дефицит дигидропиримидин дегидрогеназы
Дефицит длинноцепочечной ацил-коэнзим А дегидрогеназы
Дефицит короткоцепочечной ацил-коэнзим А дегидрогеназы
Дефицит метилентетрагидрофолат редуктазы
Дефицит прекалликреин
Дефицит синтетазы голокарбоксилазы
Дефицит фактора ХI
Иминоглицинурия
Муковисцидоз
Мышечная дистрофия Дюшенна
Нейрофиброматоз
Органические ацидемии
Первичный системный дефицит карнитина
Перманентный неонатальный сахарный диабет
Перонеальная дистрофия Шарко-Мари-Тута (часть вторая)
Перонеальная дистрофия Шарко-Мари-Тута (часть первая)
Пропионовая ацидурия
Псевдополидистрофия Гурлера
Семейная вегетативная дисфункция
Семейная гиперхолестеринемия
Серповидно-клеточная анемия (часть вторая)
Серповидно-клеточная анемия (часть первая)
Синдром Ангельмана
Синдром Бартера
Синдром Блума
Синдром Дабина Джонсона
Синдром Дауна (часть вторая)
Синдром Дауна (часть первая)
Синдром Клайнфельтера-Рейфенштейна-Олбрайта
Синдром кошачьего крика
Синдром Криглера Найара
Синдром Леша-Наяна
Синдром Лойса-Дитца
Синдром Патау
Синдром Прадера-Вилли
Синдром Эдвардса
Тирозинемия
Тирозинемия І типа
Тирозинемия ІI типа
Тирозинемия ІIІ типа
Фенилкетонурия (ФКУ)
Фруктоземия
Х-сцепленный-ихтиоз
Целиакия (часть вторая)
Целиакия (часть первая)
Церебротендинальный ксантоматоз
Цистинурия
|