


Генетические заболеванияБолезнь Фабри Болезнь Фабри (известная также как болезнь Андерсона-Фабри, дефицит альфа-галактозидазы А) является редкой наследственной Х-сцепленной рецессивной лизосомальной болезнью накопления, которая может вызвать широкий спектр системных симптомов. Болезнь названа в честь одного из ее первооткрывателей Иоганна Фабри. Патофизиология
Дефицит фермента альфа-галактозидазы А (а-GAL А, кодируется геном GLA), который возникает из-за мутации, вызывает накопление гликолипида известного как глоботриаосилцерамид (сокращенно GB3, GL-3) в кровеносных сосудах, других тканях и органах. Такое накопление приводит к нарушению их нормального функционирования. Мутации ДНК, которые являются причиной возникновения заболевания, наследуются по рецессивному Х-сцепленому типу. Болезнь поражает гемизиготних мужчин (т.е. всех мужчин), а также гомозиготных и, во многих случаях гетерозиготных, женщин. В то время как у мужчин обычно проявляются серьезные симптомы, для женщин характерна вариация течения заболевания, оно может быть бессимптомным или с проявленем тяжелых симптомов. Эта изменчивость, как полагают, связана с Х-инактивационой моделью во время эмбрионального развития женщины. Симптомы Симптомы заболевания, как правило, впервые проявляются в раннем детстве и обычно их очень трудно понять. В связи с тем, что болезнь Фабри редко встречается в практике врачей это иногда приводит к установлению неверного диагноза. Проявления болезни, как правило, с возрастом человека ухудшаются. Боль Боль во всем теле или локализованные боли в конечностях (известный как акропарестезия) или желудочно-кишечном тракте (ЖКТ) является характерным явлением для пациентов с болезнью Фабри. Считается, что акропарестезия при заболевании связана с повреждением периферических нервных волокон, передающих боль. Боль в желудочно-кишечном тракте, вероятно, обусловлена накоплением липидов в малых его сосудах, что в свою очередь, мешает кровообращению (ишемия кишечника), вызывая боль. Боль в почках
Распространенным и серьезным следствием болезни являются осложнения, связанные с почками. Почечная недостаточность (уремия) может ухудшаться со временем. Протеинурия (которая может вызвать пенистость мочи) часто является первым признаком поражения почек. Конечная стадия почечной недостаточности у мужчин, как правило, наступает в 30-40 летнем возрасте, и является частой причиной смерти. Проявления со стороны системы кровообращения Сердечные осложнения возникают при накоплении гликолипидов в клетках сердца. Влияние на сердце с возрастом ухудшается, что может привести к увеличению риска возникновения сердечно-сосудистых заболеваний. Часто у больных наблюдается гипертония (высокое кровяное давление) и метаболические изменения (кардиомиопатия). Дерматологические проявления 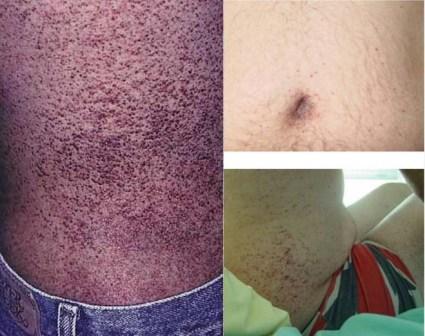 Ангиокератомы (маленькие, безболезненные папулы, которые могут появляться на любой части тела, но обычно появляются на бедрах, вокруг пупка, ягодицах, нижней части живота и в паху) тоже является распространенным симптомом. Ангидроз (отсутствие потоотделения) является распространенным симптомом в отличие от гипергидроза (повышенная потливость), который встречается значительно реже. Кроме того, у пациентов могут возникать симптомы подобные синдрому Рейно вместе с нейропатией (в частности, жгучая боль в конечностях). Поражение глаз 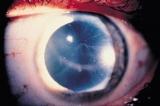 При заболевании характерны также поражение глаз, а именно помутнение роговицы (известное как кератопатия). Кератопатия может проявляться и у бессимптомных носителей, но необходимо исключить любые другие причины ее появления (например, осадок препарата в роговице). Это помутнение не влияет на остроту зрения. При заболевании характерны также поражение глаз, а именно помутнение роговицы (известное как кератопатия). Кератопатия может проявляться и у бессимптомных носителей, но необходимо исключить любые другие причины ее появления (например, осадок препарата в роговице). Это помутнение не влияет на остроту зрения.Другие симптомы включают конъюнктивитный аневризмм, заднюю субкапсунальную катаракту, отек диска зрительного нерва, отек макулы, атрофию зрительного нерва и дилатацию сосудов сетчатки. Другие проявления Другими общими симптомами заболевания являются: усталость, невропатия (в частности, жгучая боль в конечностях), цереброваскулярные эффекты, приводящие к повышенному риску возникновения инсульта, шум в ушах (звон в ушах), головокружение, тошнота, неспособность набрать вес, электролитный дисбаланс, диарея и другие проявления. Диагноз Болезнь Фабри диагностируется в том случае, если присутствуют присущие болезни симптомы, а также после анализа крови с помощью которого определяют уровень активности альфа-галактозидазы. Однако, при установлении диагноза женщинам-носителям, он может вводить в заблуждение в связи с случайной природой X-инактивации. Хромосомный анализ гена GLA является наиболее точным методом диагностики, в связи с возможностью определения многих мутаций, вызывающих заболевания. Почечная биопсия может также указать на наличие болезни Фабри, если при обследовании будут определены избыточные накопления липидов. Естественно, что альфа-галактозидаза А (а-GAL А), вероятно, будет присутствовать только на очень низком уровне в крови, особенно у мужчин. У женщин в связи с моделью Х-инактивации уровень а-GAL А, как правило, является нормальным, даже если у пациента проявляются специфические симптомы. Sifap (инсульт у молодых пациентов) - проект, в котором будут исследовать связь между инсультом и болезнью Фабри. Ошибочно установлен диагноз болезни Фабри Педиатры равно как и терапевты, часто ошибаются при диагностировании болезни Фабри. Лечение До 2000-го года, лечения болезни Фабри было направлено на преодоление симптомов, возникающих при прогрессирования заболевания. В 2001 году начали применяться три направления ферментнозаместительной терапии (ФЗТ): альфа галактозидазы (Реплагал (Replagal), производство Shire) и бета галактозидазы (Фабразим (Fabrazyme), производство Genzyme). Лечение путем замены дефицитного фермента осуществляется инъекциями каждые две недели и является наиболее применяющимся методом. Стоимость этих препаратов - высокая (примерно $ 250,000 США в год / пациента) и остается непреодолимым барьером для многих пациентов в некоторых странах. Инфузия может осуществляться самим пациентом, медсестрой дома у больного или в медицинском учреждении. Ферментозаместительная терапия не является панацеей, но может помочь нормализовать обмен веществ и предотвратить прогрессирование заболевания, а также избежать повторения симптомов. Боль при болезни Фабри тоже утоляется благодаря ФЗТ, однако схемы лечения болевого синдрома могут также включать применение анальгетиков, противосудорожных, и нестероидных противовоспалительных препаратов. Упоминания о болезни Фабри в поп-культуре: - Доктор Хаус («Полный провал», 2 серия 6 сезона (эпизод 113)) центров для пациентов с болезнью Фабри; - «Клиника» (My Catalyst, сезон 3 эпизод 12) особенности диагностики болезни Фабри. <<<
Алькаптонурия
Гистидинемия
Гомоцистинурия
Поликистозная болезнь почек
Cиндром Марфана (Болезнь Марфана)
Анемия Фанкони
Аргининосукцинатная ацидурия
Бета-талассемия
Боковой амиотрофический склероз (часть вторая)
Боковой амиотрофический склероз (часть первая)
Болезнь I клеток
Болезнь Вильсона - Коновалова
Болезнь Гирке
Болезнь Дауна
Болезнь Канавана
Болезнь кленового сиропа
Болезнь Краббе
Болезнь Ниманна - Пика
Болезнь Тея-Сакса (вторая часть)
Болезнь Тея-Сакса (первая часть)
Болезнь Фабри
Болезнь фон Виллебранда
Болезнь Хантингтона
Галактоземия
Гемоглобин Е
Гемоглобин С
Гемохроматоз
Гликогенозы
Дальтонизм (часть вторая)
Дальтонизм (часть первая)
Дефицит 3 гидрокси-метил-глутарил-КоА лиазы
Дефицит 3-метилкротонил-коэнзим А карбоксилазы
Дефицит альфа 1 антитриптисина
Дефицит бета-кетотиолазы
Дефицит биотинидазы
Дефицит дигидропиримидин дегидрогеназы
Дефицит длинноцепочечной ацил-коэнзим А дегидрогеназы
Дефицит короткоцепочечной ацил-коэнзим А дегидрогеназы
Дефицит метилентетрагидрофолат редуктазы
Дефицит прекалликреин
Дефицит синтетазы голокарбоксилазы
Дефицит фактора ХI
Иминоглицинурия
Муковисцидоз
Мышечная дистрофия Дюшенна
Нейрофиброматоз
Органические ацидемии
Первичный системный дефицит карнитина
Перманентный неонатальный сахарный диабет
Перонеальная дистрофия Шарко-Мари-Тута (часть вторая)
Перонеальная дистрофия Шарко-Мари-Тута (часть первая)
Пропионовая ацидурия
Псевдополидистрофия Гурлера
Семейная вегетативная дисфункция
Семейная гиперхолестеринемия
Серповидно-клеточная анемия (часть вторая)
Серповидно-клеточная анемия (часть первая)
Синдром Ангельмана
Синдром Бартера
Синдром Блума
Синдром Дабина Джонсона
Синдром Дауна (часть вторая)
Синдром Дауна (часть первая)
Синдром Клайнфельтера-Рейфенштейна-Олбрайта
Синдром кошачьего крика
Синдром Криглера Найара
Синдром Леша-Наяна
Синдром Лойса-Дитца
Синдром Патау
Синдром Прадера-Вилли
Синдром Эдвардса
Тирозинемия
Тирозинемия І типа
Тирозинемия ІI типа
Тирозинемия ІIІ типа
Фенилкетонурия (ФКУ)
Фруктоземия
Х-сцепленный-ихтиоз
Целиакия (часть вторая)
Целиакия (часть первая)
Церебротендинальный ксантоматоз
Цистинурия
|