


Новини генетики09 / 12 / 2010
Нейродегенеративні генетичні захворювання людини відтворені in vitro  Новий метод відстежує розвиток двох генетичних захворювань людини шляхом індукування розвитку плюрипотентних стовбурових клітин до функціональних нейронів. Дослідники з Університету Конектикут (University of Connecticut ) використовували індуковані плюрипотентні стовбурові клітини (iPS) для вивчення розвитку і патогенетичних механізмів двох дуже споріднених між собою людських захворювань. Використовуючи цей новий метод дослідники значно розширили розуміння розвитку синдромів Енгелмана (Angelman syndrome(AS)) іПрейдера-Вілі (Prader-Willi syndrome(PWS)). Новий метод відстежує розвиток двох генетичних захворювань людини шляхом індукування розвитку плюрипотентних стовбурових клітин до функціональних нейронів. Дослідники з Університету Конектикут (University of Connecticut ) використовували індуковані плюрипотентні стовбурові клітини (iPS) для вивчення розвитку і патогенетичних механізмів двох дуже споріднених між собою людських захворювань. Використовуючи цей новий метод дослідники значно розширили розуміння розвитку синдромів Енгелмана (Angelman syndrome(AS)) іПрейдера-Вілі (Prader-Willi syndrome(PWS)). Обидва захворювання є нейродегенеративними порушеннями з чіткими фенотиповими ознаками людини. Вони виникають через метилування імпритних центрів (ICs) в самій алелі, і це селективне імпритування материнської і батьківської алелей відіграє важливу роль у розвитку захворювань. Дослідники намагалися у лабораторних умовах використати iPS клітини для простеження розвитку цих синдромів в їх відповідних нейронних фенотипах для кращого розуміння механізму і розвитку захворювань. Дослідницька група з Університету штату Коннектикут встановила що батьківська UBE3A є пригнічена в функціонуванні в ASнейронів, але не в AS iPS клітинах (I) з яких були виведені нейрони.
«Насправді класним в нашій iPS моделі є те, що ми маємо цей весь процес відтворений на тарілочці (тобто в чашці Петрі)» розповідає перший автор і асистент професора генетики і біології розвитку в Центрі Здоров'я UConn (UConn Health Center) Стормі Чемберлейн. «Через те, що ми можемо переходити від стовбурових клітин до нейронів, ми можемо задати питання, а як саме це відбувається, і чи iPS-клітинна модель настільки чудово працює?» Чамберлен і її колега, Марк Лаланде (Marc Lalande), професор генетики і біології розвитку в Центрі Здоровя UConn вивчали AS і PWS на моделях мишей, але прагнуть провести кількісне метилування на людських клітинах. «Є дуже мало генетиків людини які приступали до вивчення iPS клітин, як моделі» розповідає Чамберлен «але ми можемо багато дізнатися про генетичні порушення у людини використовуючи iPS клітини». Чамберлен намагається зрозуміти як нейроно-специфічно регулюється беззмістовна транскрипція, це важливо для того, щоб пролити світло як цей ген асоціюється з AS і який це має вплив на мозок. Ген , названий UBE3A , кодує убіквітин-протеїн лігазу, яка пов’язана з деградацією протеїнів. Втрата функцій материнської успадкованої алелі UBE3A призводить до AS, і Чемберлейн потрібен метод для кількісної оцінки, як убіквітин-лігаза регулюється імпринтингом. Попередні дослідження вказують на те, що метилування може призвести до знищення перепрограмованих iPS клітин, але Чамберлейн і Леланд створили iPS клітини, які генерують функціональні AS нейрони, які ведуть IC модель. Оскільки дослідники володіють методом для вирощування AS нейронів, команда використовувала кількісний метил-специфічний ПЛР (PCR) аналіз для ідентифікування особливостей успадкування і поведінки UBE3A. Їхні дослідження вказують, що втрата функціональних алелей може бути дуже легко ідентифікована у функціональних нейронах живих організмів, які можуть допомогти розробити нові діагностичні або терапевтичні методи для хворих на AS і PWS. «Кількісний метилювальний аналіз може посприяти діагностиці, оскільки це ще простіше і більш надійніше ніж діагностика за допомогою бісульфатного метилюваного методу» розповідає Чемберлен. В кінцевому рахунку, я думаю, що ці аналізи будуть відмінно підходити для визначення інших порушень імпринтингу, які типово мають історію метилювання за типом «все-або-нічого» із доступним позитивним або негативним контролем». Стаття “Induced pluripotent stem cell models of the genomic imprinting disorders Angelman and Prader-Willi syndromes,” була опублікована у електронному вигляді 12 жовтня 2010року в журналі PNAS.
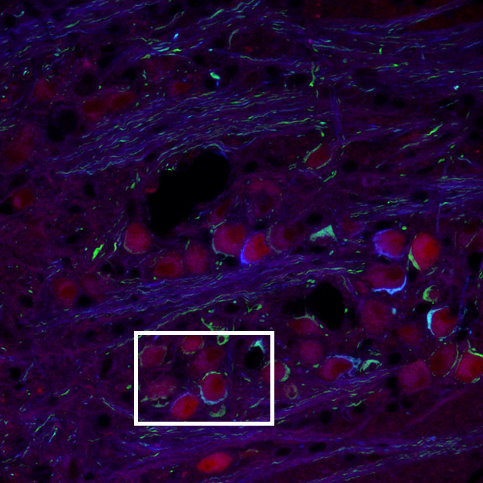
Конфокальні та двофотонні знімки слайсів зони медіального ядра трапецієвидного тіла MNTB (medial nucleus of trapezoid body) "brainbow mice", "calyx of Held", зображення отримано за допомогою Leica SP5
 <<< |