


ПубликацииСиндром Марфана: исторический ракурс и современный взгляд Синдром Марфана: исторический ракурс и современный взгляд на этиологию, патогенез, диагностику, клинику и лечение Львовский национальный медицинский университет имени Данила Галицкого Ключевые слова: синдром Марфана, этиология, патогенез, классификация, диагностика, лечение. Резюме. В обзоре литературы представлены исторические данные о синдроме Марфана с момента описания французским педиатром в 1896 году, генетические основы, классификация, патоморфология, клиническая картина трех систем организма: костно-мышечной, сердечно-сосудистой и органа зрения. Приведены диагностические критерии синдрома Марфана (Ghent nosology, 1996), дифференциальная диагностика болезней и синдромов, сходных по клинической картиной, представленны офтальмологические проявления заболевания.Перечислены основные подходы к медикаментозного и хирургического лечения сердечно-сосудистых осложнений. Синдром Марфана (СМ) - наследственное заболевание соединительной ткани с поражением скелетной, мышечной, сердечно-сосудистой систем и глаз, наследуется по аутосомно-доминантному типу. Несмотря на то, что СМ был описан более 100 лет назад, он продолжает оставаться одной из актуальных проблем медицины с позиции этиологии, патогенеза, морфогенеза, диагностики и лечения. В связи с этим до сих пор пересматривают диагностические критерии, классификацию СМ, изучаются фенотипические проявления заболевания и ведутся поиски новых методов лечения. У одного пациента с СМ может быть столько проблем со здоровьем, сколько специалистов в поликлинике.
Историческая справка. В 1896 году французский педиатр Антуан Жан Марфана описал пятилетнюю девочку с чрезвычайно тонкими и длинными конечностями, контрактурами суставов, кифосколиозом и назвал этот синдром pattes d'araignee (пальцы паука). Эта девочка могла иметь арахнодактилию с контрактурами (синдром Билса), но имя Марфана было использовано для обозначения сочетание симптомов, связанных с дефектами в гене FBN1. Позже Ашари [Achard, 1902] назвал его арахнодактилия и долихостеномелия (греч. dolicos - длинные, stenos - тонкие, melis - конечности). Существуют другие названия, которые составляют сегодня лишь исторический интерес: - синдром Марфана-Эшера; - акромакрия Пфаундлера; - акродолихия Бругшмеллера; - долихостеномелия; - синдром Марфана-Эрбе; - гиперхондроплазия Мэри и Бабоне; - акрохондрогиперплазия Валентина; - врожденная акромакрия с амиотонией Юнга; - врожденная мезодермальная дистрофия Веве; - дисмезектопия Лавал; - долихоморфия. Кроме этого, часть авторов считает, что современное определение синдрома Марфана не имеет никакого отношения к описанному Марфаном в 1896 году [Лисиченко О.В., 1986]. Поражение аорты, как симптом СМ, было выявлено только в 1943 году EtterLE, GloverLP, а роль дилатации аорты в сокращение продолжительности жизни уточнена в 1972 году MurdochJL. Роль фибриллина в патогенезе СМ описана 1990 году Холлистер ET, а также был определен локус этого заболевания хромосоме 15q21.1. Доказательство того, что мутации гена фибриллина-1 (FBN1) могут привести к СМ были описаны в 1991 году Даецом [MosheFrydman, 2008]. Синдром Марфана - редкое заболевание. Частота возникновения 2-3 случая на 10 000. Большинство авторов описывали единичные случаи заболевания.Наибольшее количество случаев описано Марфаном - 10 (1938), Грималди - 15 (1964), Маккьюсикои - 75 пробандов (1966). Отечественные исследователи диагностировали синдром Марфана у 17 больных - (Кондрашин, Неудахин, 1968); 11 - (Надарейшвили, Паламарчук, 1970); 19 - (Добро, Добро, 1970); 30 - (Гусева, 1971); 64 - (Лисиченко, 1971), 23 - (Уткин, Грушин, 1972); 10 - (Гапузов, 1973), 70 - (Семячкина, 1975) [ЛисиченкоО.В, 1986, Барашнев Ю.И. и др., 1983]. Генетика. Генетический характер СМ впервые заметил Weve H., который описал семью с несколькими больными и таким образом доказал аутосомно-доминантный тип наследования [Лисиченко О.В., 1986]. Дальнейшие генетические исследования показали, что мутация в гене фибриллина-1 (FBN1) оказывается не только у больных СМ, но при других похожих заболеваниях соединительной ткани, объединенные в группу фибрилинопатий I типа [Фищеко Я. В., 2006]. Ген FBN1 размещается на длинном плече 15 хромосомы и картирована в локусе 15q 21.1 (рис. 1). Рисунок 1. Структура пятнадцатой хромосомы человека  Примечание. 15-я хромосома человека - одна из 23 человеческих хромосом. Она содержит более 102 млн. пар оснований, что составляет 3-3,5% всего материала ДНК клетки человека. Вероятно, она состоит из 700-900 генов. Примечание. 15-я хромосома человека - одна из 23 человеческих хромосом. Она содержит более 102 млн. пар оснований, что составляет 3-3,5% всего материала ДНК клетки человека. Вероятно, она состоит из 700-900 генов. Примерно в 75% случаев заболевание передается генетически, и лишь 25% случаев заболевания вызываются спорадическими мутациями. Стоит отметить, что СМ характеризуется выраженной генетической гетерогенностью. Сегодня известно около 550 мутаций в разных семьях. Среди выраженных мутаций в гене FBN1: 57% - миссенс мутации, 18% - фреймшифт, 16% - спайс-сайт, 8% - нонсенс мутации. Зачастую при классической форме СМ характерна мутация в одном из доменов FBN1 (epidermal growth factor EGF-like domain), отвечающих за связывание кальция с фибриллином. Патологические изменения в том же локусе могут вызывать различные клинические проявления - от стертой формы с поражением одной системы организма к классической форме. Клиническое разнообразие СМ обуславливает вовлечение мутаций, локализованных в других генах, например в гене FBN2 или FBN3. Этот факт подтверждается тем, что у части пациентов с клинически выраженным СМ определяется нормальный метаболизм фибриллина, а при генетическом анализе отсутствует мутация в гене FBN1 [Ватутин Н.Т. и др., 2006, Фищенко Я.В., 2006]. Морфология. Молекулярная основа заболевания - нарушение синтеза одного из белков соединительной ткани фибриллина, который придает ей эластичность и обеспечивает сократительную способность. Основные патоморфологические изменения обнаруживаются в соединительной ткани (мезодерме), что в прошлом стало основой названия заболевания, которая сейчас не используется (dystrophia mesodermalis congenita Marfan; dystrophia mesodermalis hypoplastica).Соединительная ткань обладает повышенной способностью к растяжению и менее вынослива к физическим нагрузкам [Ватутин Н.Т. и др., 2006]. Типичные гистологические изменения в средней оболочке сосудов эластического типа, проявляются разрушением эластичного каркаса с некрозом и фрагментацией эластичных волокон, нарушением направленности и расщеплением коллагеновых волокон, дистрофией гладкомышечных клеток, накоплением между волокнистыми структурами мукополисахаридов с последующим формированием небольших кист [Смоленский В.С., 1964, Ватутин Н.Т. и др., 2006]. Классификация. Общепринятой классификации СМ не существует, однако разные авторы пытались это сделать [Лисиченко О.В., 1986]. Классификация синдрома Марфана за Лисиченко: I Форма: 1. cтертая: слабо выраженные изменения в одной или двух системах. 2. выражена: - слабо выраженные изменения в трех системах; - выраженные изменения хотя бы в одной системе; Рис. 2. Снимок черепа - выраженные изменения в двух, трех и более системах. человека, больного долихоцефалией Степень выраженных изменений:  а) легкая; б) средняя; в) тяжелая. II. Характер течения: 1. Рецидивирующее (прогрессирующее). 2. Стабильное. III. Генетическая характеристика: 1. Семейная форма (тип наследования). 2. Спорадический случай. 3. Первичная мутация Клиника. Пациенты с выраженными проявлениями СМ имеют: долихоцефалию; узкий лицевой скелет;высокий рост; низкую массу тела; длинные конечности и паукообразные пальцы; высокое небо; кифосколиоз; воронкообразную грудную клетку, а также поражение органов зрения (эктопию хрусталиков, микросферофакию; плоскую роговицу, миопию(близорукость)) . Тяжелая патология сердечно-сосудистой системы. Различают три вида сердечно-сосудистых проявлений при СМ - врожденные дефекты структуры стенок сосудов эластического типа, особенно в аорте и легочной артерии; - последствия предыдущих дефектов - аневризмы и разрывы аорты; - различные пороки развития в сочетании с СМ, например коарктация аорты, гипоплазия аорты, незаращение артериального протока [Смоленский В.С., 1964]. Аневризма аорты при этой патологии возникает с одинаковой частотой у мужчин и женщин, в возрасте 30-40 лет, с преимущественным поражением восходящей част. Имеет мешкообразный вид, с характерным поражением ее ветвей, а также изолированными аневризмами артерий [Зербино Д.Д, 2006] . Поскольку сосудистая патология при синдроме Марфана генерализованная, то поражается эластичная ткань всех сосудов. Аневризмы могут возникать не только в разных отделах аорты, но и в легочной артерии, а также в сонных, лучевых, локтевых, бедренных и других сосудах организма [Ватутин Н.Т. и др., 2006]. Рис. 3. Деформация грудины (воронкообразная грудная клетка) 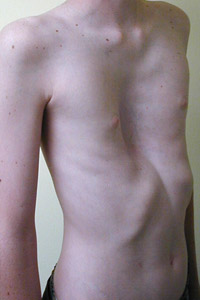 Изменения скелета встречаются у 2 / 3 пациентов, больных синдромом Марфана и включают высокий рост, астеническое телосложение, долихостеномелию, прогнатию, "готическое" небо, деформацию грудины, арахнодактилию, сколиозы, кифосколиозы, нарушение функции суставов, плоскостопие, дисфункцию височно-нижнечелюстного сустава . Изменения скелета встречаются у 2 / 3 пациентов, больных синдромом Марфана и включают высокий рост, астеническое телосложение, долихостеномелию, прогнатию, "готическое" небо, деформацию грудины, арахнодактилию, сколиозы, кифосколиозы, нарушение функции суставов, плоскостопие, дисфункцию височно-нижнечелюстного сустава . Офтальмологические проявления диагностируют практически у всех больных СМ, независимо от возраста. Чаще всего это миопия разной степени, плоская роговица, гипоплазия радужки и цилиарной мышцы, эктопия хрусталиков, изменения калибра сосудов сетчатки, косоглазие, дегенерация сетчатки, отслойка сетчатки, катаракта, глаукома. Однако два последних критерии требуют дальнейшей проверки для включения их в категорию малых критериев. Плоская роговица коррелирует с вывихом (подвывихом) хрусталиков [Ватутин Н.Т. и др., 2006, Викторова И.А., 2004, Ольхова О.В., 2010, Mema V., 2010]. Диагностика. По критериям Ghent nosology, 1996 (табл.1) СМ определяется большими и малыми клиническими признаками: в органах опорно-двигательного аппарата; органа зрения; сердечно-сосудистой системы; дыхательной системы; кожи; твердой мозговой оболочки. Для установления диагноза СМ необходимо присутствие по одному крупному критерию в двух системах и одного малого в третьей [Фищенко В. Я., 2007, Moshe Frydman, 2008]. Кроме диагностических критериев, используют фенотипические диагностические тесты СМ, а именно: - соотношение кисть / рост> 11%; - отношение размах рук / рост> 1,05; - длина среднего пальца> 10см; - отношение длины верхнего сегмента тела к нижнему <0,86; - индекс Варги <1,5 [Ватутин Н.Т. и др., 2006]. Таблица 1 Диагностические критерии синдрома Марфана
Дифференциальная диагностика. Поскольку для СМ характерным является поражение костно-мышечной, сердечно-сосудистой систем и органов зрения,то нужно проводить дифференциальную диагностику для того чтобы исключить заболевание на другие синдромы, для которых характерно поражение этих органов и систем:
- Скелетные проявления - синдром Лойса-Дитца, врожденная арахнодактилия с контрактурами (синдром Билса), синдром Стиклера, синдром Клайнфельтера гомоцистинурия, марфаноподобная умственная отсталость; - Глазные проявления - синдром Стиклера, гомоцистинурия (эктопия хрусталика), синдром Элерса-Данло, эктопия хрусталика Вайля-Марчезани, аутосомно-доминантная эктопия хрусталика, аутосомно-рецессивная эктопия хрусталика с/без эктопии радужки; - Сердечно-сосудистые проявления – синдром Лойс-Дитца, синдром Марфана 2 типа, пролапс митрального
 клапана, МАSS фенотип (семейный пролапс митрального клапана), кистозный медиальный некроз Ердгейма с расслоением восходящей аорты, семейная аневризма аорты, двустворчатый аортальный клапан с расслоением восходящей аорты [ Барашнев, Кадурина, Лисиченко, Umamahesh, Моше Фридман]. клапана, МАSS фенотип (семейный пролапс митрального клапана), кистозный медиальный некроз Ердгейма с расслоением восходящей аорты, семейная аневризма аорты, двустворчатый аортальный клапан с расслоением восходящей аорты [ Барашнев, Кадурина, Лисиченко, Umamahesh, Моше Фридман]. В 2004 году Т. Mizuguchi (и др.) сообщили, что причиной СМ может быть нарушение обмена трансформирующего фактора роста β (ТGFRB2). Бельгийские ученые Барт Лоец и пел. в 2006 году сообщили, что мутации в TGFBR1 является причиной аневризмы аорты и марфаноидного фенотипа (Loeys-Dietz синдром).Хотя расслоения аорты является общим для обоих синдромов (Марфана и Лойс-Дитца), в последнем, эктопии хрусталика является редким симптомом, а у пациентов имеются гипертелоризм (90%), волчья пасть или язычок (90%) и завитки артерий (84%) .
Прогноз. Если не производить лечение болезни Марфана, то в 50% случаев поражения сердца и сосудов приводят к смерти при отсутствии хирургического вмешательства. Наиболее опасной является прогрессирующая дилатация кольца аортального клапана и расслоение аорты. Однако, за последние 19 лет госпитальная летальность при плановых операциях в Национальном институте сердечно-сосудистой хирургии имени Н.Н. Амосова в Украине составила 0%, при 164 прооперированных. Своевременная кардиохирургическая коррекция сердечно-сосудистых проявлений и осложнений синдрома Марфана позволила увеличить продолжительность и улучшить качество жизни многих пациентов с СМ [Хорошковатого О.В., 2010].
Лечение. В лечении СМ используют медикаментозное и хирургическое лечение, одинаково улучшают продолжительность жизни больных до 60-70 лет. Медикаментозная терапия должна быть продолжена, и после операции. Среди медикаментов применяют β-блокаторы, которые снижают скорость расширения аорты и могут увеоичить продолжительность жизни пациентов. Целью лечения должен быть строгий контроль над артериальным давлением (систолическое давление до 120 мм рт.ст. и 110 мм рт.ст. для пациентов с расслоением аорты). Большинство центров мира используют именно β-блокаторы (бисопролол). Блокаторы рецепторов ангиотензина II являются потенциально полезными, поскольку они приводят к антагонизму ТGF-β. Клинические испытания, которые проводятся сейчас, показывают хорошую эффективность лозартана для профилактики аневризмы аорты у больных СМ (ESC Guidelines, 2010).
Сегодня существует три основных подхода к хирургическому лечению сердечно-сосудистых осложнений у больных СМ: - во-первых - протезирование восходящей аорты и аортального клапана с использованием кондуита, который содержит клапан (операция Bentall de Bono); - во-вторых - отдельное протезирование аортального клапана и восходящей аорты в случае, если синусы Вальсальвы не изменены, но наблюдаются выраженные деструктивные изменения в створках аортального клапана; - в-третьих - выполнение клапано-сохраняющих операций в случае интактности аортального клапана или умеренно выраженных морфологических его изменений. Литература: 1. Барашнев Ю.И., Казанцева Л.З., Семячкина А.Н., Бухны Л.Ф. (1983) Дифференциальный диагноз болезни Марфана и некоторых сходных с ним синдромов (синдром Билса и Стиклера) у детей // Вопросы охраны материнства и детства, № 4. С. 41-46.
2. Белоконь Н.А., Кубергер М.Б. (1987) Болезни сердца и сосудов у детей: Руководство для врачей. В 2 т. Т. 2. – М.: Медицина, – 480 с.
3. Богомолов Л. И., Фисанович Т.И., Карлова Т.Ф. (1982) Поражение периферических артерий при синдроме Марфана // Клиническая хирургия, № 7. С. 66.
4. Ватутин Н.Т., Склянная Е. В., Кетинг Е.В. (2006) Синдром Марфана // Кардиология, № 1. С. 92-98.
5. Викторова И.А., Нечаева Г.И.(2004) Синдром Марфана в практике терапевта и семейного врача: диагностика, тактика ведения, лечение, беременность и роды // Русский медицинский журнал, №2.
6. Демин А.А., Антонов О. С., Семенова Л. А., Сентякова Т.Н., Карчова Т. Ю., Муранова Г. В., Рыскинд В.А., Кузнецов В.А. (1985) Синдром Марфана: полиморфизм клинических проявлений // Терапевтический архив, т. 57, № 4, С. 133-135.
7. Дмитриев В. И., Марченко А. М., Марин А. И. (1981) Особенности клиники синдрома Марфана в юношеском и зрелом возрасте // Врачебное дело, №7, С. 78-80.
8. Зербіно Д. Д. (2006) Патологія аорти: класифікація, хвороби і синдроми, проблеми етіології // Медицина транспорту України, № 2, С. 6-14.
9. Кадурина Т. И., Горбунова В. Н.(2009) Дисплазия соединительной ткани. Руководство для врачей. – СПб.: Элби – СПб. – 704 с.
10. Контридзе В. С., Кванталиани И. Г. (1980) Три случая трансплантации склеры при синдроме Марфана // Вестник офтальмологии, №4. С. 68-69.
11. Кравченко І.М., Сітар Л.Л., Федонюк Л.Я., Захарова В.П.(2007) Аневризми висхідної аорти та аортальна недостатність при синдромі Марфана: проблеми хірургічного лікування та морфології // клінічна анатомія та оперативна хірургія, №4, С. 58-61.
12. Кузьмінський А.П., Малярська Н.В., Опалинський Ю.А. (2008) Гострі порушення мозкового кровообігу і синдром Марфана // Буковинський медичний вісник, № 3. С. 123 – 125.
13. Лазовскис И. Р.(1981) Справочник клинических симптомов и синдромов. 2-е изд., перераб. и доп. – М.: Медицина, - 512с.
14. Лайбер Б., Ольбрих. Г. (1974) Клинические синдромы. М.: Медицина. – 477с.
15. Леванюк В.Ф., Тидир А. А. (1983) Диагностическая ценность индекса телосложения Варги при синдроме Марфана // Клиническая медицина, т.61, №9. С. 131-134.
16. Лисиченко О. В.(1986) Синдром Марфана. – Новосибирск: Наука, – 164с.
17. Ольхова О.В. (2010) Родинна форма синдрому Марфана: варіанти вад очей // Мистецтво лікування, №6 (72), с.110 – 112.
18. Оспанова Л.С., Вятчинин Н.Г., Турлубеков К.К.(1986) Синдром Марфана // Здравоохранение Казахстана, №10. С. 70-71.
19. Смоленский В.С. (1964) Болезни аорты . – М.: Медицина. – 283с.
20. Фіщенко В. Я., Фіщенко Я. В. (2007) Семіотика синдрому Марфана. Укр. мед. альманах, 10(2): 172–175 с.
Автор: Зербино Д.Д., Ольхова О.В., Жураев Р.К www: Источник <<< |
||||||||||||||||||||||||||||||